A Not so Non-Infectious Disease
As James Gillray depicted back in 1799, it turns out that “tiny devils” might play a role in gout after all.
Introduction
Two misconceptions currently distort our understanding of gout. The first is that gout is a disease of modern origin. It is not, and even though it is more prevalent now than ever, it has been shown to have occurred as far back as the time of the dinosaurs.1)https://www.nature.com/articles/387357a0
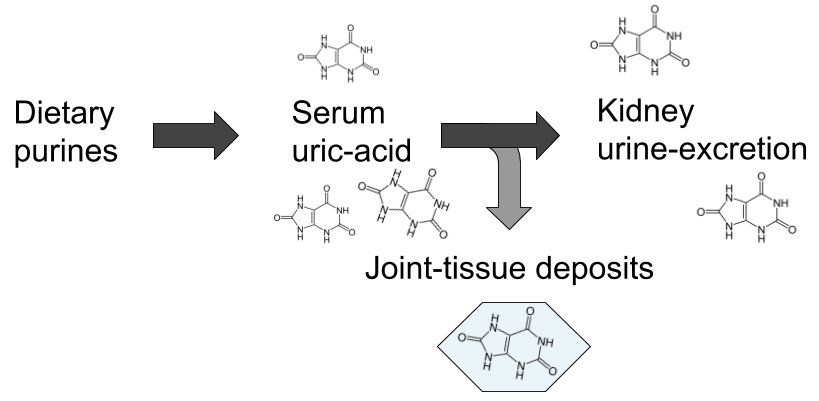
The second, and maybe largest misconception is that it is exclusively a non-infectious metabolic disease. The conventional explanation goes something like this: over-consumption of food containing high amounts of a class of molecules called purines leads to rising levels of uric acid in the blood when they are metabolised, and when serum levels surpasses the point of saturation, which is often referred to as hyperuricemia, uric acid crystallises to monosodium-urate (MSU) that then gets deposited in the articular tissues causing inflammation and severe pain. Purines can be said to be the back-bone in DNA-molecules, and are also part of the cellular energy-carrier ATP, hence they are essential parts of all living cells, making them very strange dietary candidates for disease.
However, although I believe the above patho-mechanical model to be largely erroneous, there is some truth to the connection with hyperuricemia and purines, but not in the way most people think as we shall see later on in this article. Gout is still the common example brought up to distinguish disease of metabolic origin from those caused by infection in the medical literature, and it is often spoken of as the most well-studied disease of the former category.2)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5512152/ This is strange, and also a bit ironic considering that the only experimental model that has been able to prove a causal pathway involves microorganisms, while metabolic models have consistently failed. This means that current treatment regimes, heavily rooted in the belief of a metabolic cause, are potentially very misguided, which would explain the apparent dissonance between our perceived high degree of understanding of gout pathophysiology and the inferior outcomes of corresponding clinical endeavours.3)https://www.ncbi.nlm.nih.gov/pubmed/22863577 Maybe we are too heavily influenced by past assumptions, and are coming at the problem from the wrong perspective.
Failure of Hyperuricemic Models.
The presence of MSU-crystals in the joints of patients with gout has been conclusively demonstrated. However, the explanation as to how and why they end up there has largely been based on assumptions rather than clinical evidence. As a matter of fact, when put to the test experimental models starting from the assumption that hyperuricemia results in tissue deposits of MSU-crystals has repeatedly failed to demonstrate this. The most common model of hyperuricemia is a mouse-model with genetic knock-out of the gene coding for an enzyme called urate-oxidase (UOX) which is synthesised by most mammal species and converts uric acid into a more water-soluble molecule called allantoin when it reaches the liver. Shutting down this pathway results in greatly elevated levels of serum uric acid but none of the clinical signs of gout such as articular deposits of MSU and formation of tophi.4)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC43025/?page=5 This is true both in long- and short-term experimental scenarios.5)https://www.ncbi.nlm.nih.gov/pubmed/28729031 The only place where MSU-crystals are observed in this model is the kidney, causing mechanical blockage in the renal tubuli, resulting in death in a large part of the animals within a few weeks. The kidneys are the main route for uric acid elimination, which probably explains this phenomenon.6)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3359272/ But no other tissue deposits are observed in hyperuricemic models, thus disproving the notion of uric acid “spill-over” from the blood, and instead points vaguely towards a mechanism of insufficient local tissue clearance.
The Physiology Behind Hyperuricemia.
Hyperuricemia is still the variable with the strongest clinical association to gout, with a massive 6- and 13-fold increased relative risk in hyperuricemic men and women respectively.7)https://arthritis-research.biomedcentral.com/articles/10.1186/s13075-018-1697-6 However, an apparent paradox is that only a small minority of patients with hyperuricemia go on to develop gout. 8)https://academic.oup.com/rheumatology/article/49/11/2010/1785765 This probably means that hyperuricemia is a precondition, or simply an epiphenomenon, but that it is in and of itself insufficient to cause the disease.
Reasons for the Relative State of Hyperuricemia in Humans.
Humans, along with other higher primates and also species of birds and reptiles have a loss of function-mutation on the UOX-gene, resulting in an almost 10-fold higher level of serum uric acid in us, compared to other mammals.9)https://academic.oup.com/mbe/article/33/9/2193/2579393 This does not seem to be an error, but a feature brought about through natural selection with advantages such as less need for water and dietary sodium, improved mechanisms for conservation of nutrients and energy (like nitrogen recycling and easier conversion of dietary fructose into fat), and it is also thought to compensate for a previous loss of function-mutation in the primate-lineage related to endogenous synthesis of vitamin-C. 10)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3203212/11)https://pubmed.ncbi.nlm.nih.gov/2575132/12)https://www.ahajournals.org/doi/full/10.1161/01.HYP.0000028589.66335.AA13)https://www.pnas.org/content/111/10/376314)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5512146/
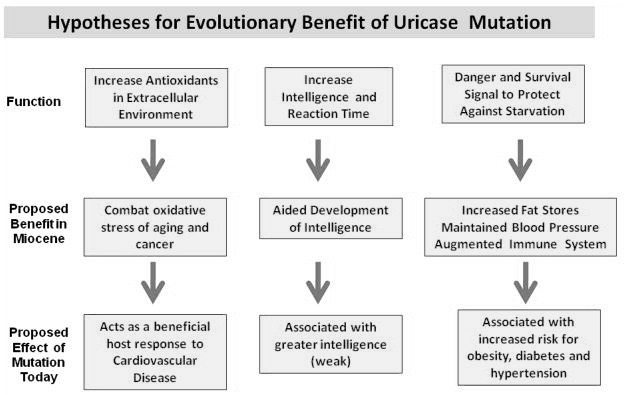
There also seems to be a relationship with energy-metabolism per unit of mass and the need for a functioning UOX-gene because of differences in ATP-turnover, explaining why this mutation was possible in humans, presenting an almost 40-fold difference compared to smaller mammals, where the same mutation has proved to be fatal.15)https://www.frontiersin.org/articles/10.3389/fphar.2019.00098/full#B26 The loss of function-mutation of the UOX-gene in theory predisposes us to develop gout through limiting the window between homeostatic levels and hyperuricemia, although this seems to be accounted for by a big overcapacity in renal excretion.16)https://www.ncbi.nlm.nih.gov/pubmed/20627967/
Relationship to the Metabolic Syndrome.
Hyperuricemia is not an exclusive marker for gout, and seems to be a common finding in many other diseases such as cardiovascular disease and metabolic syndrome.17)https://www.sciencedirect.com/science/article/abs/pii/S002191501931421218)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5941695/ This was previously believed to be because of insulin resistance, but a 2017-paper investigating the temporal relationship between hyperuricemia and insulin resistance showed that hyperuricemia does not follow insulin resistance, but rather precedes it, implying reverse causation.19)https://www.ahajournals.org/doi/full/10.1161/hypertensionaha.117.09508 Increased fat-mass is probably not the cause either, and although the increased amount of fat mass has been shown to be a potent source of uric acid, this relationship has been questioned by a recent study showing similar serum uric acid levels between lean and obese patients with metabolic syndrome.20)https://www.jbc.org/content/early/2013/08/02/jbc.M113.485094.full.pdf21)https://www.ncbi.nlm.nih.gov/pubmed/31016168 The relationship between Body-mass-index (BMI) and hyperuricemia is also not linear, and instead seems to be U-shaped, with significant risk-increase in those with low BMI, as well as in those with a high value.22)https://www.ncbi.nlm.nih.gov/pubmed/31767029 Hyperuricemia is also not to be considered a universal marker for disease. For example, in neurodegenerative diseases there seems to be an inverse relationship with uric acid, where a history of gout even seems to confer protection from disease such as Parkinsons and Alzheimers.23)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5233635/ There has been speculations of hyperuricemia being a underlying cause of metabolic dysfunction, but recent studies are starting to show that this is probably not the case.24)https://www.ncbi.nlm.nih.gov/pubmed/31876630
A Marker for Cell-Death.
Specifically, hyperuricemia seems to be a marker for cellular energy distress and cell death (trough increased breakdown of ATP and DNA), explaining diverging direction of association in different pathophysiological contexts.25)https://www.frontiersin.org/articles/10.3389/fphar.2019.00098/full26)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6348815/ In line with this, serum uric acid has proven to be a marker of illness-severity associated with poor prognosis in sepsis.27)https://www.hindawi.com/journals/ijn/2015/301021/ In gout, this sets up a potential scenario where local energy-distress and cell death inside of the joint-tissues, for reasons that will be discussed later, results in increased formation of uric acid that can’t be delivered to the circulation (in contrast with the conventional theory that uric acid is deposited in the tissues) at a sufficient rate, perhaps partly because of a weakened chemical gradient as a result of hyperuricemia. But the question remains as to how and why this happens and why it results in full-blown clinical gout. Fortunately, other experimental models might hold the answer.
Mycotoxins, a Missing Piece of the Puzzle?
So far, only one experimental model has been able to successfully replicate the full clinical picture seen in gout, and it has largely, surprisingly, been overlooked. The model originated from the observation that domestic chickens, raised for food-production are commonly seen to develop articular gout when they eat mouldy corn, and in a study from 1981, researchers confirmed the causative agent to be oosporine, which is what is called a mycotoxin.28)https://www.ncbi.nlm.nih.gov/pubmed/7329919
A Response to Fungivorism.
Mycotoxin are a class of secondary metabolites produced by some fungal species in response to being eaten.29)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3790476/ For the fungi, this is not only a defence-mechanism, and it is thought to have evolved as a way for the fungus to use insects and other fungivorous species as a source for carbon and nitrogen, essentially killing and digesting them from within, a mechanism that then also seems to out-compete bacteria and other rival microorganisms in claiming the remaining carcass, that is then used for reproduction through sporulation.30)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6126850/31)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5745424/32)https://www.ncbi.nlm.nih.gov/pubmed/2402370533)https://www.ncbi.nlm.nih.gov/pubmed/28193896 In line with this, most toxins produced by fungi directed towards single-celled organisms (such as bacteria) as defence are secreted, while those that are directed towards fungivores are instead stored in the fungal cells and are taken up during predation.34)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6126850/
Mycotoxins Attack the Mitochondria.
Mycotoxins seems to work mostly through disruption of cellular energy-homeostasis by attacking mitochondria and their DNA, causing cell-death through energy deficiency, resulting first in subtle clinical loss-of-function symptoms. 35)https://www.ncbi.nlm.nih.gov/pubmed/679706736)https://www.ncbi.nlm.nih.gov/pubmed/2699755537)https://www.ncbi.nlm.nih.gov/pubmed/28401999 This theoretically manifests first in the body’s top energy-consuming organs such as the brain, liver, kidneys and muscles, and later in less metabolically active tissues, such as the joints. This results in increased metabolism of endogenously derived purines in the body coming mostly from DNA and ATP released by dying cells, ending up as uric acid, first in tissues and then in the blood, later to be excreted. As previously mentioned, the kidneys are greatly affected by exposure to mycotoxin due to its high rate of ATP-turnover, making some early researchers to label it as being primarily a nephrotoxin.38)https://www.ncbi.nlm.nih.gov/pubmed/7092745 However, there are some data showing normal serum levels of uric acid and increased renal uric acid excretion in a great proportion of patients experiencing an acute gout flare, which seem to infer normal kidney function, at least intermittently.39)https://www.ncbi.nlm.nih.gov/pubmed/2197749040)https://www.ncbi.nlm.nih.gov/pubmed/30345643 Also, in the aforementioned studies of avian gout the disease could not be attributed to the co-occurence of neither hyperurecemia nor renal failure.41)http://journals.mu-varna.bg/index.php/bmr/article/viewFile/221/221Early studies of patients with gout have also shown normal kidney-excretion of uric acid in response to an oral purine-load.42)https://academic.oup.com/jn/article-abstract/106/3/428/4763582 Epidemiologically, kidney-disease seems to infer about a two-fold increase in relative risk of gout, and having gout is associated with about 30 % increased risk of developing kidney-disease in multivariate analyses.43)https://bmjopen.bmj.com/content/5/4/e00684344)https://bmjopen.bmj.com/content/9/8/e031550
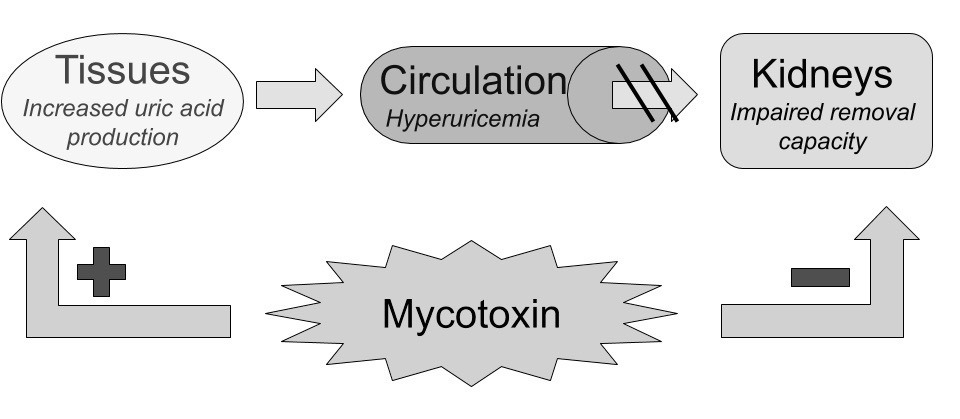
Diversion of Uric Acid Towards the Intestines.
Impaired kidney-function lowers the rate of uric acid clearance through urine, diverting it towards the bodys second option of elimination; the intestines. This mechanism has been proven by nephrectomy models, showing maintained serum levels of uric acid and a reciprocal increase in intestinal excretion.45)https://link.springer.com/article/10.1007%2Fs10157-013-0806-846)https://bmcvetres.biomedcentral.com/articles/10.1186/s12917-019-1886-9 Hyperuricemia might occur as a result of uric acid production overwhelming both renal and intestinal removal pathways, however if these removal pathways function normally increased uric acid production might also be masked because there is no reciprocal increase in serum-concentration which might explain normouricemia in some cases of gout (more on this further on in the article).
Intestinal Tissues Themselves are a Potent Source of Uric Acid.
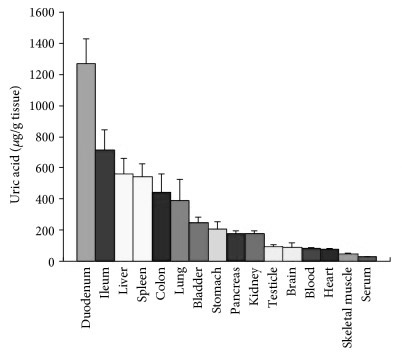
The lining of the small intestine is also exceptionally high in uric acid, which is probably because uric acid is one of the body’s major endogenous danger-signals corresponding to cell damage, making the immune-system very reactive to the event of an intestinal barrier breach, to protect against systemic infection. 47)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739491/48)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648987/ Rapid recruitment of immune-cells seems very appropriate when also considering the apparently large microbial appetite for uric acid.49)https://www.ncbi.nlm.nih.gov/pubmed/2575132 Increased cellular turnover in the intestine, which could potentially be a direct consequence of infection or mycotoxin-exposure to the intestinal lining, can increase uric acid content both in the blood and in the intestinal lumen which in the later might end up contributing to microbial growth and virulence. Studies have shown a hyperuricemic response to gastrointestinal infections with a wide range of pathogenic instigators, in particular, infection with Shiga-toxigenic E. coli has been shown to cause a spike in serum uric acid to the point of MSU-formation in the kidneys, perhaps as a consequence of intestinal injury. Crystals also appear in the gut lumen as a direct response to infection with this particular pathogen, probably because of leakage of extracellular DNA by host cells.50)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4807499/
The Consequence of Increased Microbial Access to Nitrogen.
The end result of all this is an increased amount of uric acid both in circulation, tissues and most importantly, in the intestinal lumen, the suspected place of fungal colonisation. Ammonia, which can be easily produced by microbial breakdown of uric acid is one of the main preferred sources of nitrogen by most microorganisms, including fungi, and when ammonia is available they selectively repress uptake of less preferable nitrogen containing molecules, a mechanism known as nitrogen catabolite repression.51)https://www.ncbi.nlm.nih.gov/pubmed/257513252)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5777862/ The fungi then uses the nitrogen in large part for de-novo purine synthesis, which is a mechanism that for the most part seems to be evolutionary conserved between fungi and humans, so much so that for example fungal yeasts are considered to be appropriate models to study human purine-related disease.53)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6356901/ Nitrogen availability in the setting where tissues are dominated by fungi also enable them to synthesise even more mycotoxins, thus potentially creating a vicious circle once they get a good grip on the intestinal lining.54)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4246892/ A controversial study in mice showed that intestinal colonisation of germ-free mice with the common beer-yeast Saccharomyces cerevisiae caused an increase in host purine metabolism and excretion of uric acid through the intestine, exacerbating symptoms of colitis, an effect which seemed to be dependent on uric acid, implying increased microbial virulence in connection with nitrogen availability.55)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5994919/ Also, a recent experimental study using the UOX-KO model have shown that hyperuricemia induces microbial breach of the intestinal barrier. Together, these two studies lends theoretical support to the hypothesis of increased microbial virulence in the presence of excessive endogenously derived uric acid in the intestinal lumen.56)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6755192/ This hypothesis might also explain why body retains most of the uric acid that is excreted through the small intestine before it reaches the colon, which is the place of greatest microbial abundance in the gastrointestinal tract, and instead diverts most of it towards the main route of excretion; the kidneys.57)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5739491/ The intestines might provide a great buffer-system for scenarios of temporary renal overload, or it might also be the case that the body uses the microbiome to recycle uric acid to ammonia which it then re-absorbs and uses for de-novo protein-synthesis.58)https://www.ncbi.nlm.nih.gov/pubmed/257513259)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5512146/ This mechanism has been extensively studied in ruminant animals and is the reason why they can survive on their naturally low-nitrogen diets.60)https://biomedres.us/pdfs/BJSTR.MS.ID.003401.pdf
The Human Body is Normally Inhospitable to Fungi.
Humans are normally very resistant to intestinal colonisation by fungi. This is partly because of the presence of the bacterial microbiome, making the human intestine largely uninhabitable to fungi because of fierce competition for resources. They are also vastly outnumbered, and it is estimated that only around 0.01 – 0.1% of the total number of intestinal microbes are fungal species, although studies has yet to control for total biomass.61)https://www.ncbi.nlm.nih.gov/pubmed/31216461 Most bacterial species also have the ability to destroy both fungi and their mycotoxins, and on top of this the fungi also need to outsmart the host immune-system, which is probably the main limiting factor, and explains why fungal infections in particular are more common in immunocompromised patients.62)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5553267/63)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5834427/64)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5724762/65)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3155160/
Viral Cause is Also Possible.
Several viral outbreaks in chicken-farms has also been shown to lead to gout in infected animals, but MSU was predominantly found in the ureters and intestines, and only in some of them was it found in joints.66)https://www.ncbi.nlm.nih.gov/pubmed/24015918 Viruses are obligate intracellular parasites, and hence require the cellular metabolic machinery of the host to reproduce. The process of viral replication causes an increase in the synthesis of DNA and also increased cellular turnover, mimicking the pathophysiology of tumors, and when the immune-system starts to intervene by attacking viral-infected cells, this might free up large amounts of purines, resulting in an increased amount of uric acid in tissues and in the circulation.67)https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-019-0678-968)https://www.frontiersin.org/articles/10.3389/fmicb.2019.02314/full Since the cells in joint-tissues have a slow rate of metabolism, these tissues might not provide the best setting for a viral pathogen, perhaps explaining why joint-MSU was a rare finding in the before-mentioned avian viral outbreaks. Currently only about 1% of cases with acute articular inflammation is suspected to be of viral origin, but this might also be an underestimate.69)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4868140/
Dietary Considerations.
Sugar and Alcohol.
One method that fungi uses to reduce competition from bacteria is production of ethanol from simple sugars essentially claiming the nutrient-pool for itself.70)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4262006/ This is very interesting considering the two main dietary risk-factors for gout are intake of alcohol and sugar-sweetened beverages, which are also very low in purines (with the exception of beer). 71)https://www.ncbi.nlm.nih.gov/pubmed/3106101872)http://apjcn.nhri.org.tw/server/APJCN/27/6/1344.pdf73)https://www.ncbi.nlm.nih.gov/pubmed/19353717 The impact of sugar-sweetened beverages on the risk of gout is usually, and also wrongfully, blamed on fructose, and consumption of fruit-juices even seems to carry a slightly lower risk in comparison, however the risk is still significant, and around 80 % higher than in people who don’t regularly consume fruit-juice, while fruit intake in contrast seems to be associated with a minor risk reduction.74)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3368949/75)https://www.ncbi.nlm.nih.gov/pubmed/31061018 Increased intake of sugar and alcohol might also explain the seasonal pattern of incident gout, being more common in the summer-months.76)https://www.ncbi.nlm.nih.gov/pubmed/28283767
Yeast-Based Foods.
Most fungi can be considered to be predominantly hitch-hikers in the human gastrointestinal tract, merely passing through, and although colonisation has been proven to occur, it is still rare for previously mentioned reasons. However, this does not mean that hitchhiking fungi are harmless, and it is possible for them to claim the gastrointestinal content instead of host tissues making them able to survive the transit through the whole length of the gut while remaining virulent. This makes a “fungal elimination-diet” potentially very effective. Some data in support of this comes from experimental feeding-trials and shows that when purposefully avoiding to consume food that contains fungal yeasts, such as beer and bread (which are based on the yeast s.cerevisiae, one of the suspects in clinical gout, either directly or indirectly through symbiotic mechanisms), the prevalence of fungi in stool gets reduces to zero within days.77)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5874442/ A study in humans has also shown increased levels of uric acid in the blood when using single-celled yeasts as a dietary protein-source.78)https://www.ncbi.nlm.nih.gov/pubmed/5460023 Hence, it might be wise to avoid foods that contain yeast all together if you have gout.
Meat and Fish.
Intake of meat and fish is often cited as major dietary risk factors for gout due to the high purine and protein (another source of uric acid) content in these foods, but when comparing the highest versus the lowest quintiles of intake in the population, the increase in relative risk is very small at around 30%.79)http://apjcn.nhri.org.tw/server/APJCN/27/6/1344.pdf This means that if you belong to the fifth of the population that consumes the highest amount of meat, you have an absolute risk of getting gout of about 5%, compared with 4% in the general population.80)https://www.health.harvard.edu/arthritis/are-you-at-risk-for-gout This seems disproportionally small when viewed from the perspective that excess dietary purines causes gout, especially when compared to the risk associated with intake of purine-free food items such as sugar sweetened beverages which is associated with more than a 200 % increase in relative risk.81)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6502023/ However, the small association between meat and fish intake and gout is probably not just coincidental, and seems to be quite consistent across multiple studies. The data is however possibly confounded by protein intake, which is associated with a similar increase in relative risk of around 30% when comparing the first and forth quartiles, but the relationship does not seem to be dose-dependent.82)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4939435/ One hypothesis is that meat and fish provides gut microbes with an easily accessible nitrogen source when eaten in excess, which is probably more relevant in the case of already established disease. Purines are great food for fungi, as is glutamine and ammonia which is also present in great quantities in these food-items.83)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5488104/84)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3249386/85)https://www.ncbi.nlm.nih.gov/pubmed/473559686)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5777862/ In a study following patients with diagnosed gout for a year, it was shown that total purine intake was associated with a 476 % increased risk of experiencing a gout flare when comparing the highest versus the lowest quintiles of intake, controlling for medications and alcohol intake. When stratifying patients according to intake of animal protein, the corresponding increase in risk was 220 %.87)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3889483/ It is however very unlikely that high intake of meat and fish, or protein in general causes gout when considered as independent variables, as nitrogen alone is insufficient to cause fungal growth in the human gut.
Vegetables and Dairy.
Another hole in the conventional purine-centered perspective is that intake of protein- and purine-rich vegetables seems to be slightly negatively associated with gout, and an apparent paradox is that vegans, that supposedly avoid the main dietary culprits of gout such as meat and fish, have been shown to have higher serum uric acid levels when compared to meat-eaters.88)http://apjcn.nhri.org.tw/server/APJCN/27/6/1344.pdf89)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3572016/ Dairy-products which are usually high in protein is associated with a risk reduction of almost 50%.90)http://apjcn.nhri.org.tw/server/APJCN/27/6/1344.pdf This risk reduction, and also that associated with vegetable intake is probably due to the fact that both plants and dairy-products tend to harbour lactic acid-producing bacteria which are fierce competitors to fungal moulds.91)https://jfoodprotection.org/doi/pdf/10.4315/0022-2747-33.12.55092)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5553267/
Purine and Protein Intake Syncs With Renal Excretion.
Systemic purine or protein overload through dietary intake is unlikely to cause gout and the hypothesis is full of holes and inconsistencies. In the healthy state the body is actually striving more towards retention of uric acid rather than elimination, reflected by the fact that around two thirds of circulatory uric acid is of endogenous origin and that about 90% of the uric acid that gets excreted by the kidneys are reabsorbed.93)https://emedicine.medscape.com/article/241767-overview#a494)https://www.ncbi.nlm.nih.gov/pubmed/20627967 This is because uric acid serves important functions (even though its not essential) in the human body, as previously discussed, which is why it literally thinks twice before getting rid of it.95)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648987/ The process of successive pseudogenization in primate evolution also echoes this very strongly96)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3956161/ Should the amount of dietary purines or protein become to great, the amount excreted goes up reciprocally by fine-tuned signalling pathways.97)https://www.ncbi.nlm.nih.gov/pubmed/629476598)https://www.ncbi.nlm.nih.gov/pubmed/7424639 Thus a purine-rich diet only gives a small, and transient increase in serum uric acid is in and of itself insufficient to cause either hyperuricemia or gout.99)http://www.jrheum.org/content/jrheum/29/7/1350.full.pdf
Micro-Ecology is the Potential Link Between Diet and Disease.
It is not how the food gets metabolised in the body that mediates the dietary associations with gout, but rather how the food we eat interacts with the bacterial and fungal microbiome. To cause gout we essentially need to eat as if we were purposefully growing fungi in our gut, which would be a diet consisting first and foremost of large amounts of alcohol and simple carbohydrates, and then easily accessible amino acids from sterile food-items such as heavily processed meat. To be successful in our attempt to grow intestinal fungi, our diet must also simultaneously be extremely low in foods preferred by commensal bacteria such as vegetables and dairy. Except for directly consuming food or beverages containing fungi or fungal metabolites, it is most likely impossible to cause gout by just doing one of the before-mentioned things, like eating lots of meat, or avoiding vegetables.100)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5512154/ Fungi require just the right environment to grow and produce mycotoxin.101)https://www.intechopen.com/online-first/growth-of-fungal-cells-and-the-production-of-mycotoxins It really is a disease that is the sum of its parts, and when considering gout as being primarily caused by fungi, the connections to dietary risk factors starts to make sense, explaining previous paradoxes related to the purine-centered model.102)http://apjcn.nhri.org.tw/server/APJCN/27/6/1344.pdf
Another Perspective on Obesity.
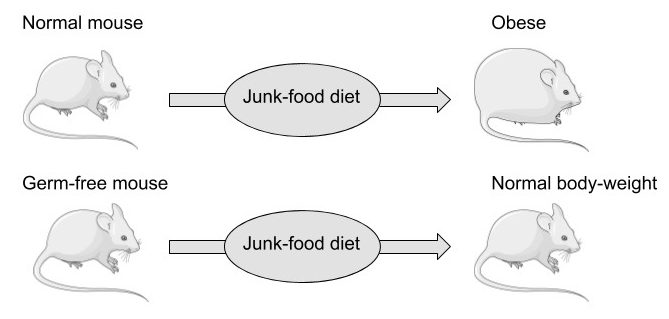
Another diet-related factor that should be mentioned again at this point is obesity, which is very strongly and dose-dependently associated with gout, with those having a BMI around 40 also having an almost 5-fold increase in relative risk compared to that of people within the normal range.103)https://www.ncbi.nlm.nih.gov/pubmed/25209031 This is however, contrary to popular opinion, probably not because of the increase in fat-mass but might instead simply reflect an increasingly large caloric intake from mostly processed food, increasing the available nutrients to the microbiome. Nutrients don’t simply diffuse from the intestinal lumen into the blood when food is consumed, they are actively absorbed, and there is strong evidence showing that obese people have a higher absorptive capacity in the small intestine compared to lean people.104)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5609829/ This might reflect the fact that the body is in direct competition with the gut microbiome for intestinal nutrients that are of mutual interest (such as glucose, fats and amino acids), and that quick absorption and high storage-capacity (fat-mass) are favourable consequences of this arms-race, reducing the amount of nutrients available to intestinal microbes and hence lowering their inflammatory potential. This notion is supported by studies showing that mice don’t seem to have a need to absorb and store excess body-fat from a hyper-caloric diet in the absence of an intestinal microflora, which also might explain the “obesity paradox”, the observation that increased fat-mass seems to be protective from cardiovascular disease in those with metabolic syndrome.105)https://www.ncbi.nlm.nih.gov/pubmed/17183312106)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6503652/
However, the process of weight-loss is still favourable to gout patients, and has been shown to reduce the frequency of acute gout flares, but it is probably more a consequence of diminished caloric intake rather than reduced body-mass.107)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6125106/
Non-Dietary Considerations
General Health and Immune-Function.
As previously mentioned, the virulence of fungi is heavily dependent on host immune-function, and fungi are not considered to be invasive in the healthy state, meaning that factors that are associated with general health might also be protective in relation to fungi-related pathologies by maintaining a strong immune system.108)https://www.ncbi.nlm.nih.gov/books/NBK8103/ For example, a proper sleep-pattern has shown to be associated with reduced levels of serum uric acid, which probably mirrors tissue resilience in relationship to environmental factors.109)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6521014/ Unfortunately, the inverse is also true, and people with a weakened immune-system such as patients subjected to immunosuppressant therapy have a greater risk of acquiring fungal disease.110)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3155160/ Immunosuppressive treatment is mandatory for patients receiving an organ-transplant to avoid graft-rejection, and research show that treatment with a common immunosuppressive drug called cyclosporin causes severe hyperuricemia in four out of five patients and also gout in more than 10%, which is more than double the absolute risk for the disease in the general population.111)https://www.ajmc.com/journals/supplement/2005/2005-11-vol11-n15suppl/nov05-2216ps435-s442. Coincidentally, cyclosporin is also a fungal metabolite, making it very evident that some fungal species actively target host immune-functions.112)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3630736/
Antibiotics.
Common antibiotics, such as penicillin have been shown to cause gout, and these substances were also originally produced by fungi. 113)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5512154/
Diuretics.
Another class of drugs related to gout are diuretics, which causes retention of uric acid.114)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3253199/ The mechanism behind this seems to be related to water-loss and dehydration, which is also an independent risk factor gout.115)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6125106/ An intriguing thought is that since fungal biomass contain a large percentage of water, they might also have ways to exploit host hydration.
Oral Hygiene.
Oral hygiene may also play a part, and studies show that tooth-brushing can greatly reduce levels of common pathogenic fungi in the stool.116)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5874442/ The mouth seems to be a particularly good niche for the fungi to occupy in the setting of a junk-food diet because of large and frequent delivery of simple sugars and carbohydrates in combination with very limited local absorptive capacity in the host, making frequent between-meal tooth-brushing particularly important in this scenario.117)https://www.ncbi.nlm.nih.gov/pubmed/23799070 Patients with gout also exhibits signs of oral microbial dysbiosis with increase prevalence of pathogenic bacteria related to periodontitis.118)https://www.ncbi.nlm.nih.gov/pubmed/30231244
Reversal of Chronic Gout.
When trying to induce remission of chronic gout, we have to think in reverse. How do we design a diet and related lifestyle factors to eliminate a fungal pathogen that has gotten a good grip on the digestive tract? In theory we need to create an intestinal ecosystem that is more inhospitable to fungi. When looking at the common dietary risk factors it gets very evident where to start, and that is to eliminate simple sugars and alcohol from the diet completely, together with a reduction in energy intake. Apart from sobriety, a good staring point for the sake of simplicity might be a very low-carbohydrate, paleo-style diet in combination with intermittent fasting, which would in theory starve the fungi, who are heavily dependent on a frequent flow of preferred nutrients to remain virulent. Including dairy in the diet, as well as a variety of non-starchy vegetables might also be of benefit through getting help from various other gut-commensals to fight of the fungi. Its also important to avoid direct exposure to fungal molds, both from food and from other sources in the environment.
Exercise Might be Counterproductive.
Strenuous, physical exercise, which is usually portrayed as a universal cure for every disease may be detrimental in the context of gout. This is hinted by a study showing an slight increase in serum uric acid by around 5 % after a 12-month exercise intervention compared to a control-group.119)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4120269/ This might be because of sweating and dehydration, but there is also mechanistic research showing that strong physical exertion increases the degradation of purines to uric acid in local tissues as a response to energy-depletion and mechanical stress which might constitute a problem, as I will explain further on in this article.120)https://www.frontiersin.org/articles/10.3389/fphar.2019.00098/full Exercise is in and of itself not health-hazardous, quite the contrary, and the increased endogenous uric acid is excreted in the urine or recycled back into the tissues, but in the pathophysiological context of gout, it is probably counterproductive.121)https://www.frontiersin.org/articles/10.3389/fphar.2019.00098/full There is also reason to limit or avoid specific exercise of previously gouty joints, if the underlying life-style related factors have not been dealt with, as we shall se in the section below.
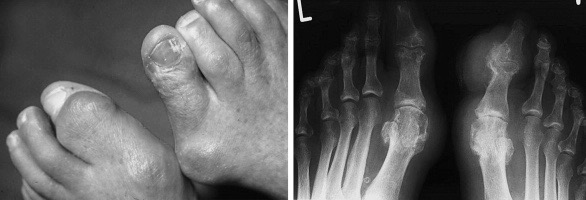
The Big(-Toe) Question.
This is perhaps the most talked-about mystery surrounding gout. Why the big toe? Gout seems to have a near exclusive preference for peripheral joints such as those of the hands and feet (and less commonly the knees and elbows), almost completely sparing joints close to the core such as hips, shoulders and the spine. The initial presentation is usually mono-articular, involving only one joint, but may over time progress to become oligo- or poly-articular. However, there seems to be a very striking predilection for the first metatarsophalangeal joint, the first joint of the big toe, and the reason for this also shines light on two local tissue factors associated with articular gout. The first factor being mechanical stress and damage to articular tissues, which increases local purine turnover and hence also the amount of uric acid inside the joint, and the second factor is related to circulation and temperature which influences uric acid removal and solubility.
Biomechanical Stress.
The connection to biomechanical stress and tissue turnover is shown by the strong relationship between the process of osteoarthritis and the location of MSU-crystals. A study examining 7855 human talar-bones from 4007 cadavers found that in 92 % of cases with evidence of articular gout (which was very rare, at around 5%) there were also gross signs of cartilage degeneration, with crystals being located to areas of the bone evidently subjected to great biomechanical stress.122)https://www.ncbi.nlm.nih.gov/pubmed/18412302/ Increased tissue turnover might not be the only factor linking gout with osteoarthritis, and its possible that pre-existing joint-inflammation might contribute indirectly to MSU-formation by impairing the flow of uric acid from the joint space back to the circulation because of swelling of synovial tissues.123)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4372016/ The direction of causality seems to be that osteoarthritis predisposes to gout in the affected joint, rather than the other way around.124)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3117776/ Hence, the predilection for the big toe from a purely biomechanical perspective might be because of the striking prevalence of osteoarthritic changes in this joint, which is around 40% in middle-aged to older adults.125)https://www.ncbi.nlm.nih.gov/pubmed/20472083/ However, this does not explain why gout is almost absent in other joints where degenerative changes are common such as the spine, and why its fairly common in the ankle where osteoarthritis is rare.
Temperature and Circulation.
This might be resolved by considering the more distal and peripheral anatomical localisation of joints and other tissues commonly hit by gout and their proximity to the skin, which makes them more exposed to changes in external temperature. In the setting of local hypothermia the solubility of uric acid is reduced, increasing the chance of MSU-formation.126)https://www.ncbi.nlm.nih.gov/pubmed/5027604/ Cold exposure may also further affect uric acid clearance by causing local vasoconstriction and reduced micro-circulation.127)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4843861/ Local thermal and vascular conditions is probably the main reason why tophaceous gout outside of peri-articular tissues commonly shows up around the olecranon and the earlobe.128)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3168008/ Inferior circulation in joint-tissues in general might also explain why MSU form in joint-tissues, and why it does not seem to happen in muscle-tissue which is highly vascular. Reduced micro-circulation may also be a another factor linking gout with the metabolic syndrome, considering the increased prevalence of peripheral artery-disease in this population.129)https://www.ncbi.nlm.nih.gov/pubmed/7712701130)https://www.jvascsurg.org/article/S0741-5214(06)00411-3/fulltext
The Connection Between Micro-Crystals and Inflammatory Pain.
This combination of increased uric acid formation through mechanical stress, reduced solubility by lowered temperature and reduced rate of elimination through insufficient circulation sets up the necessary local precondition, allowing systemic factors such as mycotoxin-mediated cytotoxicity and hyperuricemia to tip the scale towards uric acid saturation and crystallization in the peripheral joint tissues. However, there are a few more variables that need to be addressed to fully explain uric acid crystallization and initiation of the following inflammatory response. The problem is that this is an area that has been severely under-researched over the last three decades, so we have to resort to a greater level of speculation.131)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4606994/
A Third Way of Uric Acid Removal.
An interesting observation is that the final steps in MSU crystal-growth are driven by the body itself through production of MSU-specific antibodies. These antibodies bind to preformed crystals and enhance the process of MSU-formation from soluble uric acid.132)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4606994/ The reason for this is not yet fully understood, but might be a way to make uric acid immunologically active, priming it for removal through phagocytosis by macrophages and neutrophilic cells, hence constituting a third way of uric acid elimination.133)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648987/ This might be a physiological buffer-system, and there is some data indicating that phagocytic removal of uric acid might be non inflammatory in the healthy state.134)https://onlinelibrary.wiley.com/doi/pdf/10.1002/art.10614 However, if the amount of MSU passes a threshold where tissue homeostasis is severely threatened, the inflammatory response, which is essentially a chain of events that causes a stronger and more rapid recruitment of additional immune-cells to the area, might be triggered which is hinted by the large difference in total MSU-volume observed between symptomatic and asymptomatic hyperuricemia.135)https://www.ncbi.nlm.nih.gov/pubmed/25637002/ The immunological cell-count in the tissues might get so high that it is reaches levels normally only found inside of septic joints.136)https://www.ncbi.nlm.nih.gov/pubmed/25102511 Removal of MSU through phagocytosis is extremely slow in established gout, and it has been shown to take months to years before the crystals has disappeared from the joints.137)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1954685/
Mechanism of Inflammation.
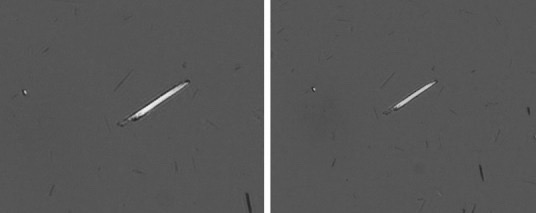
It was early suggested that the needle like structure of MSU-crystals explained their inflammatory and nociceptive potential, but this was recently refuted by a study comparing articular injections of either amorphous or needle-shaped MSU, showing no difference in their ability to cause inflammation and pain. Instead, the study confirmed the present scientific consensus, that it is not the crystals themselves that trigger pain by direct chemical stimulation of nociceptors, but that pain is caused indirectly through pro-inflammatory signalling by phagocytes that has ingested MSU, and this in turn activates these receptors in the joint tissues.138)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5895116/
The pro-inflammatory activity in tissue-resident phagocytes also seems to increase in response to systemic pro-inflammatory signals coming from outside of the joints, like IL-17 that is secreted by T-cells, which has been shown to be greatly elevated in the serum of patients experiencing an acute gout flare, subsequently causing phagocyte secretion of IL-1B, which is the primary driver of the inflammatory response seen in gout arthritis.139)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4292791/140)https://www.frontiersin.org/articles/10.3389/fimmu.2019.00070/full141)https://www.ncbi.nlm.nih.gov/pubmed/29476737 Coincidentally, IL-17 is also a chief systemic signal in the case of fungal infections, adding to the clues pointing towards gout being a fungal disease.142)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4426252/
Using NETs to Trap Microcrystals.
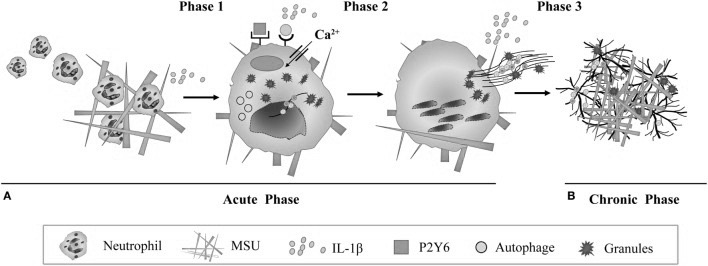
Neutrophils, which are a class of phagocytes seen in large numbers at sites of formed MSU, are shown to deploy what is called neutrophilic extracellular traps (NETs), which is a mechanism that these cells use to physically trap invading microbes but has recently also been shown to be involved in the removal of micro-crystals. This happens by neutrophils first attempting to phagocytose a crystal, and then dissolving its own membrane, leaking its DNA which then forms a sticky, web-like structure, trapping the crystals, possibly for larger phagocytes like macrophages since neutrophils themselves are smaller than MSU-crystals 143)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5361057/ It is unclear weather MSU-triggered NETs are solely a mechanism to trap crystals, or if its also a trigger-mechanism to use NETs for protection against invading microbes. Some authors also speculates that NETs might be exploited by pathogens.144)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4807499/ Interestingly, macrophages that has consumed NETs are found in the synovial fluid of gouty joints and have long been called “Reiters cells” because that they were first observed in Reiters syndrome, which is another disease where a systemic infectious stimuli causes local articular inflammation.145)https://onlinelibrary.wiley.com/doi/pdf/10.1002/art.10614146)http://www.annclinlabsci.org/content/47/3/253.full Over time the formation of these NETs leads to encapsulation of MSU and is thought to be one of the main mechanisms later involved in cessation of inflammation and the appearance of tophi, which is thought to be a consequence of failed phagocytic removal.147)https://rmdopen.bmj.com/content/1/Suppl_1/e000046
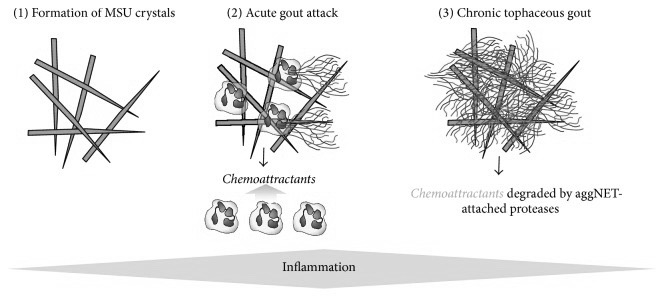
Danger-Signals.
The inflammatory nature of MSU seems to be more than just a simple question of tissue saturation of uric acid, and both MSU-formation and inflammatory activity might be influenced by microbial signals. As previously mentioned uric acid is a potent danger-signal in the body corresponding to cell death and tissue damage. Damage does not usually constitute a significant threat to the body in and of itself, since it possesses innate mechanisms for cell division and repair. What does however signify a great threat is the simultaneous signalling of tissue injury and microbial breach, which means that the immune system need to get in a tug-of-war with microbes over spilled nutrients that is otherwise safely confined on the inside of the skin and mucosal barriers, to avoid fuelling microbial virulence. This might be the reason why simply injecting antigens (traces of endogenous tissues or cells) into tissues do not usually cause an immune response in the absence of co-stimulation from a microbial adjuvant.148)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648987/
The “Double-Hit” Hypothesis.
There are a couple of lines of evidence pointing towards that this is also the case with MSU in connection to gout. A study published in 1974 showed that molecular traces of dead bacteria (endotoxin), either directly injected together with MSU into the joints, or given orally after Sterile MSU-injection caused clear signs of pain, compared with injection of the same dose of MSU without endotoxin which caused no pain.149)https://onlinelibrary.wiley.com/doi/pdf/10.1002/art.1780170415 According to observations from the same study, endotoxin also seem to posses a high degree of chemical attraction to MSU-crystals (hinting at the potential antimicrobial purpose of MSU-triggered NETs), which means that immune-cells in gouty joints might get a “double-hit”, receiving both an antigen and an adjuvant, when attempting removal through phagocytosis, explaining their increased inflammatory activity, and also the severe pain that is associated with gout. It is also of note that this study used a much lower articular dose of MSU when compared to more recent studies that manages to induce articular inflammation from MSU in the absence of an adjuvant, which might help explain cases where tissue MSU remains asymptomatic.150)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5895116/
Connection with Microbial Dysbiosis.
More support for the double hit-theory comes from an experimental study showing that injection of MSU into the joints of mice reared in a completely sterile environment caused substantially less inflammation and pain when compared with conventionally raised mice, An effect that was shown to be dependent on the presence of bacterial metabolites in the blood.151)https://onlinelibrary.wiley.com/doi/full/10.1002/art.39107 In line with this, there is also strong evidence linking gout to dysbiosis of the gut microbiome, and stool microbial profile measured by PCR has proved to be more accurate in the diagnosis of gout than using serum uric acid.152)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4757479/ Like previously discussed, it is possible that hyperuricemia also contributes indirectly to increased microbial dysbiosis and breach of the intestinal barrier, giving various microbes access to the the circulation and thus also to peripheral tissues, and since uric acid can be metabolised and used as a nitrogen source by most fungal and bacterial species they might be very attracted to gouty joints.153)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3646786/
Injuries are Ideal Locations for Pathogens.
Increased purine metabolism associated with mechanical injury in a joint might in and of itself also be a risk of infection. The pathway of purine de-novo synthesis is strikingly similar in humans and single-cell organisms, making injured tissues ideal locations for pathogens to try and occupy, especially when also considering the increased delivery of nutrients to injured tissues.154)https://www.ncbi.nlm.nih.gov/books/NBK2414/155)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6356901/ In support of this, gout and other pre-existing joint diseases seems to predispose to septic arthritis, and several cases of local joint co-morbidity with gout and septic arthritis have been reported in the literature.156)https://www.ncbi.nlm.nih.gov/pubmed/26170377157)https://ard.bmj.com/content/56/8/470158)https://www.ncbi.nlm.nih.gov/pubmed/12730521/159)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6404712/ There are also some studies that found traces of bacteria in the synovial fluid of gouty joints. 160)https://www.ncbi.nlm.nih.gov/pubmed/10618069161)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1829110/ Areas of high tissue-turnover in general are also great niches for pathogens to occupy, and the most loaded joint in the body during daily activities such as walking is the ankle-joint, which is also the most common location for gout after the first metatarsal-phalangeal joint.162)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3117776/ After the ankle, the the joints that are subjected to the greatest level of peak biomechanical stress during walking are those of the knee and hip, which in turn are the most common locations affected by septic arthritis.163)https://link.springer.com/chapter/10.1007/978-1-4939-0745-8_8164)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4228814/
Conclusion.
So, a more accurate explanation for articular gout might go something like this: a disturbance in the intestinal ecosystem favours species of fungi capable of producing mycotoxin (or other microbes, through a similar mechanism). A state of systemic cytotoxicity ensues when mycotoxin dissolves in the blood, causing a great increase in cell-death and thus also of endogenous purine turnover resulting in rising levels of uric acid in the tissues. Among the first tissues to be affected are the kidneys, which lowers their capacity for uric acid excretion leading to increased serum levels, which in turn causes a weakened chemical gradient for uric acid removal from tissues via the circulation. Local tissue factors in peripheral joints, such as high level of biomechanical stress, insufficient circulation and low temperature further exacerbates the problem of regional uric acid formation in relation to clearance, resulting in tissue supersaturation. Normal removal pathways still being largely overwhelmed, MSU-crystals starts to form in concert with local immune-cells priming them for removal by phagocytosis, causing inflammation and pain that is possibly further amplified by signs of microbial breach of mucosal barriers.
Essentially, gout is caused when we behave in a way that is congruent with the will of a mycotoxin-producing fungi, providing it with its favourite nutrients and eliminating the biological competition from other members of the microbiome, and also from our own immune system by adopting corresponding dietary habits and lifestyle factors. Weakened defences and an overly abundant supply of resources are ideal conditions for parasites, of any kind. A strong immune-system and a diet thats is in accordance with host biology will instead set the stage for mutualistic exchange with other organisms, leading not just to freedom of disease but also to greater health through cooperative effort.

Hudiksvall, Sweden
- Tendinopathy, A Mechanical Problem With a Metabolic Root - 3 June, 2021
- The Ecology of Over-Nutrition - 7 January, 2021
- What Happens When You Inhibit Intestinal Fat-Absorption? - 19 November, 2020
References
Be First to Comment