
A Mechanical Problem With a Metabolic Root
Poor health is a slow killer of tendons, and while dead tendons make for fantastic bow-strings, they make terrible muscle-attachments
After centuries of scientific inquiries you’d think that at least some of the many mysteries surrounding chronic musculoskeletal disease would be solved by now, and among those would definitely be the painful tendon. But no.
Introduction
Its label has been continually changing with changing opinions about the associated histopathology, mostly going back and forth between “tendonitis” and “tendinosis”, highlighting its inflammatory and degenerative characteristics respectively.1)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6045756/ Since no firm scientific consensus was reached on the dominant pathophysiological nature of the condition in this regard, the term tendinopathy (TP) emerged, which simply means “tendon-disease”, or “we sort of still don’t really know what we are dealing with”.
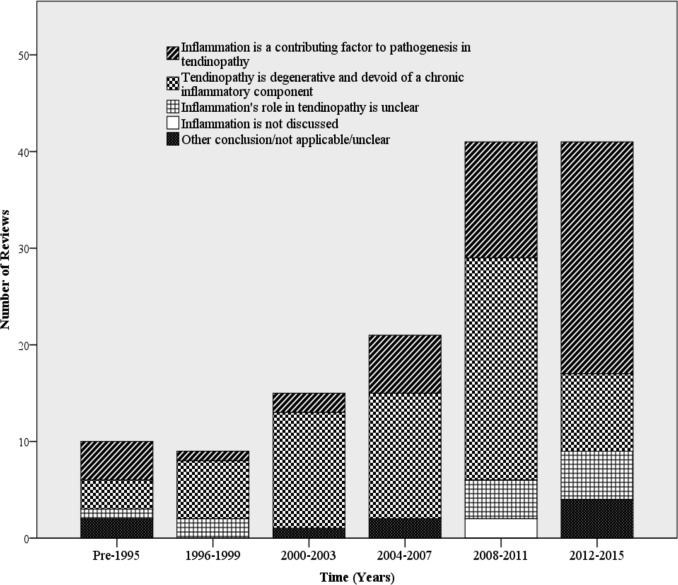
However, as I will try to explain in this section, I believe that this ambivalence can be resolved by viewing inflammation and degeneration as two inter-related phenomenons with a common root-cause.
In the literature, TP is defined both as a clinical diagnosis and as a histological entity, or as a combination of the two. This sets up for some potential errors when comparing studies. To add to this problem, clinical TP is sometimes defined as a self-reported condition by the patient without any input from a medical professional, and the different methods used to objectively define histological TP have varying degrees of sensitivity and specificity. Also, there is a dissonance between tissue-pathology and clinical symptoms, with a large portion of those with the histopathological change consistent with TP being asymptomatic which means that studies using either definition alone are prone to statistical errors. Studies also generally focus on one or a few anatomical locations when mapping out different correlations, which is also a major weakness of the current literature. This inevitably set the stage for some potential confusion going forward, but I’ll try my best to avoid it.
A basic premise in this section is that TP is a singular condition, expressed differently depending on location and circumstance, and that histological change precede and predispose to clinical symptoms, and constitute an independent risk-factor for morbidity.
Assigning a superior importance to “structure” is currently a very unpopular stance in the academic world surrounding musculoskeletal disease, where chronic pain is increasingly being framed as being a maladaptive neuropsychological phenomenon. However, I believe that this is the consequence of not knowing the nature of the problem well enough, and that such explanations is what our minds drift towards by default when our logic seem to fail us.
Disappointing Results of Modern Clinical Interventions
It should go without saying that without a proper definition of a problem, or its nature, the chances of finding a working solution are minuscule at best. Therefore it should come as no surprise that the results of commonly applied treatment-protocols used over the past 30 years have proven to be rather unimpressive. The results of three large meta-analyses of TP at different anatomical locations including between 29 to 42 different treatments unanimously show that the effects of most interventions are comparable with each-other, and that individually they barely beat waitlist-controls and placebo-arms in clinical trials.2)https://bjsm.bmj.com/content/55/5/2493)https://pubmed.ncbi.nlm.nih.gov/31171514/4)https://pubmed.ncbi.nlm.nih.gov/33148599/ This probably means that natural history and nonspecific effects of care contribute with a majority of observed “effects” of treatment, which means that to patients with persistent symptoms they wont help much at all. This is probably also a major reason why the subjective experience of patients with chronic TP is characterised by frustration and resignation, because nothing really helps in a meaningful way and no one seems to know why.5)https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0233459
Tendinopathy is a Prerequisite to Tendon-Tears
Many clinicians wrongfully define TP only as a problem of subjective nature, that there is nothing wrong with the tissue, and that the patients can safely engage in exercise as long as pain-levels are low. This notion seem to be supported when looking at a study where a clinical diagnosis of TP was proven to be associated with a negligible increase in relative risk of tendon-tears of about 4%.6)https://pubmed.ncbi.nlm.nih.gov/28540301/ However, in sharp contrast, there is a very strong relationship between a histologically confirmed diagnosis of TP and incident tendon-ruptures which was confirmed by a large study showing that 97% of ruptured tendons displayed histological signs of TP compared with about 34% of control-tendons which means that practically all of such injuries are preceded by TP (the remaining 3% were caused by things such as intratendinous tumours and foreign bodies) and that a large part of the asymptomatic population is at risk.7)https://pubmed.ncbi.nlm.nih.gov/1748700/ Another study also concluded that while both ruptured tendons and tendons affected by TP were more degenerated than control-tendons, ruptured tendons where significantly more histologically degenerated than un-ruptured tendons only affected by TP.8)https://pubmed.ncbi.nlm.nih.gov/11740288/ Degeneration therefore seems to be a true prerequisite to tendon-rupture, which is also supported by studies on basic science which show that healthy tendons are twice as strong as muscle-tissue, and tensile loading of musculotendinous units will cause rupture in the muscular part before it can injure the tendon.9)http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3312643/10)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5328946/ One study on mice also showed that the tensile strength of tendons affected by TP where halved in compared with healthy tendons.11)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7709031/ This frames TP as a problem of gradual degeneration, where pain is just a tip of the ice-berg (that does not always float above sea-level, such as for asymptomatic/histological TP). But the question is: what then cause tendons to degenerate?
Not a Simple Problem of Overload
Because of the high prevalence of TP in athletic populations and in groups with physically demanding occupations, it has long been thought to be a simple problem of mechanical overload.12)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3435934/13)https://doi.org/10.1080/15438627.2017.139374814)https://pubmed.ncbi.nlm.nih.gov/2213764/ This type of reasoning might be somewhat applicable in sub-groups of extremely active individuals as cross-sectional studies in such populations have shown an exponential positive relationship between weekly hours of training and a clinical diagnosis of TP where the prevalence was found to be 9-fold in participants who reported training for more than 20 hours a week compared with a reference-group that trained less than 2 hours per week.15)https://pubmed.ncbi.nlm.nih.gov/28151759/ However, there are also many people that suffer TP without participating in any physically demanding activity which complicates the before-mentioned conclusion of TP being mainly a mechanical problem.16)https://www.tandfonline.com/doi/full/10.1080/09638280701785825 Also, cross-sectional data tells us very little about causation, and looking instead to prospective data, a different picture emerges. In pooled analyses of prospective studies there are usually either no, limited or conflicting associations between physical activity-measures and TP, both in heterogenous populations and in athletes specifically, which seems very strange from the point of view that TP is a problem caused by “overuse”.17)https://bjsm.bmj.com/content/53/21/135218)https://www.sciencedirect.com/science/article/abs/pii/S1440244019305997 The same results are also seen in occupational settings, and a systematic review on the contribution of work-related mechanical variables have concluded that there is insufficient to very limited scientific support for them having a causal relationship with TP, and interestingly a pooled analysis of prospective occupational cohorts showed that a higher cardiometabolic risk-score were among the few variables that survived multivariate analysis while factors such as work-load, BMI and job-satisfaction failed to persist.19)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7857538/20)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5450050/
Associations to Markers of Metabolic Health
Following this trail further, there is another group that have comparable rates of TP-prevalence to athletic populations while instead being more on the opposite side of the activity-spectrum, and that is patients with type-2 diabetes mellitus (DM2). A histopathological diagnosis of TP is among the strongest internal prospective risk-factors of symptomatic/clinical TP by a factor of 4.0-7.3, and a study has shown that the prevalence of such findings are 55% in patients with DM2 (mean age of 60) compared with 19% of matched controls, and a large meta-analysis have also concluded that TP is 3,7 times more frequently occurring in those with DM2.21)pubmed.ncbi.nlm.nih.gov/27633025/22)https://pubmed.ncbi.nlm.nih.gov/33500797/23)https://pubmed.ncbi.nlm.nih.gov/26598716/ Interestingly the rate of histologically confirmed TP in the achilles tendon found in diabetics is similar to the rate found in a small study of asymptomatic runners, at about 45%.24)https://www.ncbi.nlm.nih.gov/pubmed/31261019
The Effects of Body-Mass
The association between DM2 and TP is commonly thought to be confounded by the mechanical consequence of obesity which is a prevalent characteristic in diabetic populations, and this in combination with a general deconditioning effect of inactivity is often thought to explain the prevalence of TP in sedentary populations.25)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5738911/ However, this has recently been refuted by a recent systematic review of risk-factors associated with TP in a weight-bearing tendons such as the achilles which showed that neither BMI nor the level of physical activity were significant predictors prospectively.26)https://bjsm.bmj.com/content/bjsports/53/21/1352.full.pdf Contrastingly, another recent systematic review that included mostly retrospective-, case control- and and cross-sectional studies did show a large and significant correlation between tendinopathy and BMI, and looking at the available data on achilles-TP there was a 3.8- and 6.6-fold increased prevalence in class I-II and class III obesity respectively.27)https://doi.org/10.1097/corr.0000000000001261 In the same study, obesity was also shown to be associated with a 1.9-fold increased risk of TP at non-weight-bearing sites such as the common tendon of the forearm-flexors. Together this seems to suggest two things. First that obese people who develop TP tend to do so more commonly in areas subjected to increasing levels of mechanical stress, and second that the relationship between obesity and TP-prevalence is not purely mechanical at its root and therefore perhaps related to other features commonly associated with obesity such as impaired metabolic health. Interestingly, in a recent large multivariate analysis (using data from nearly 17 million people), hypercholesterolemia (high LDL-C) was shown to be related to TP and tendon-rupture in the achilles-tendon in both under- and over-weight patients, and actually significantly more so in the former compared with the latter which further suggests that BMI is mainly a modulator of and not a cause of TP.28)https://pubmed.ncbi.nlm.nih.gov/34676271/
Impaired Metabolic Health is an Independent Risk-factor
There are multiple lines of evidence in support of an independent effect of metabolic health on TP prevalence and incidence. Starting with experimental studies, induction of DM2 either by diet or through non-dietary means have been shown to cause TP both in weight-bearing and in non weight-bearing tendons.29)https://www.ncbi.nlm.nih.gov/pubmed/3064694730)https://www.ncbi.nlm.nih.gov/pubmed/2990781131)https://pubmed.ncbi.nlm.nih.gov/28095484/32)http://www.ncbi.nlm.nih.gov/pubmed/2548954333)https://www.ncbi.nlm.nih.gov/pubmed/24360194/ Several studies on human subjects have shown a strong association between TP and unfavourable serum lipid-profiles in physically active individuals and in diabetics.34)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5436990/35)http://www.mltj.online/wp-content/uploads/2019/09/Coombes.pdf In a three year long prospective study of 5856 individuals (including both diabetic patients and non-diabetics, mean age 62 years) with varying levels fo physical activity, those with a HbA1c within the pre-diabetic range and above had about three times higher risk of tendon injury in the lower extremity, and individuals with metabolic syndrome had a 2.5-fold risk of TP in both the upper and lower extremity.36)https://pubmed.ncbi.nlm.nih.gov/33963621/ Looking at runners, an unfavourable metabolic profile with elevated HbA1c, triglycerides, total-cholesterol and reduced HDL proved to be significantly associated with achilles-TP independently of BMI and milage.37)https://pubmed.ncbi.nlm.nih.gov/30367279/ A pooled analysis of prospective occupational cohorts (mentioned previously) showed a fold-increase in risk of TP in a non weight-bearing tendon (the common tendon of the wrist-extensors) of between 2.8 and 6.2 (depending on the mode of evaluation) in workers with a higher cardiovascular risk-score, an association that was also independent of work-load.38)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5450050/ Another prospective study found a 6-fold increase in risk for shoulder-TP in those with an unfavourable cardiovascular risk-score, and this relationship was also found to be dose-dependent.39)https://pubmed.ncbi.nlm.nih.gov/28002354/ Looking at studies on tendon-tears, after smoking the greatest risk-factor was found to be dyslipidemia which gave a 5-fold increase in risk compared with controls, and a history of cardiovascular disease has been found to be the most significant predictor of severity.40)https://pubmed.ncbi.nlm.nih.gov/26321466/41)https://pubmed.ncbi.nlm.nih.gov/32309364/42)https://www.ncbi.nlm.nih.gov/pubmed/21138564/ Rates of re-tearing after surgery have been shown to be significantly higher in patients with DM2 compared with controls by a factor of 2.3 and diabetic patients also display inferior recovery to non-diabetics at 6-month post-operative follow-ups.43)https://pubmed.ncbi.nlm.nih.gov/33225006/ There is also experimental evidence supporting a connection metabolic disease with tendon healing-capacity and mice have been shown to display an inferior healing-response to tendon-injury after induction of diet-induced obesity.44)https://www.ncbi.nlm.nih.gov/pubmed/28686669/45)https://www.ncbi.nlm.nih.gov/pubmed/24658034/
A False Dichotomy
Inflammation is now widely accepted as being the mechanism that drives metabolic disease, and it could also be what mediates the connection between TP and declining metabolic health. If true, this would explain why TP also seems to be a frequent co-morbidity in other inflammatory diseases.46)https://doi.org/10.1080/09638280701785882 As mentioned in the introduction to this section, there is a longstanding debate on weather TP is an inflammatory or degenerative condition, and I believe that this conflict could be resolved now by the fact that the current literature clearly shows that both features are present.47)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7006114/ Importantly, like I explained in my article on the pathophysiology of osteoarthritis there is probably no reason to further differentiate the two, especially since they seem to be causally linked to each other with chronic low-grade inflammation leading to degeneration by impairing tissue-maintenance and repair.48)https://pubmed.ncbi.nlm.nih.gov/31838495/
-Se my article on inflammation for a more thorough explanation of this concept-
A Weak-Spot in the Vascular Infrastructure
To understand how inflammation and degeneration are linked in TP we first need to understand how tendons are nourished. Historically, tendons were long considered to be a “dead tissue” that lacked both a cell-population and a vascular supply. Now it is widely known that there are cells residing inside tendon-tissue and that they also interact with the vascular system. However, because of the significant amount of strain transmitted in this tissue, tendons cant afford to have penetrating blood-vessels and they therefore have to use a similar strategy as joints for sustaining an adequate exchange of nutrients and metabolites which is by diffusion to and from a superficial peripheral vascular supply. This mechanism works differently in tendons that are covered with a tendon-sheet from those who are not, similarly to how the vascular infrastructure varies with different types of joints. Sheeted tendons are structured like synovial joints where nutrients are supplied to the tendon by formation of synovial fluid synthesised by the lining cells of the tendon-sheet (paratenon) that then works its way in towards the central part of the tendon through diffusion and convection (convection is the flow of fluids, generated in tendons as well as in cartilage by cyclic mechanical loading, which is essential for nutrient to reach the more central/deep parts of such tissues illustrated by in-vitro experiments showing degeneration when it is absent49)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8142054/). Unsheathed tendons are supplied in a manner similar to osteochondral joints and also rely on diffusion and convection of nutrients, but from a direct vascular supply that surround individual fascicles of the tendon. In practice both these supply-routes have the same weak-spot in that small disturbances at the vascular interface can have profound consequences for nutritional exchange, especially for the more central parts of the tendon which is usually where degeneration starts when the vascular supply is disturbed.50)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4650849/51)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC128932/
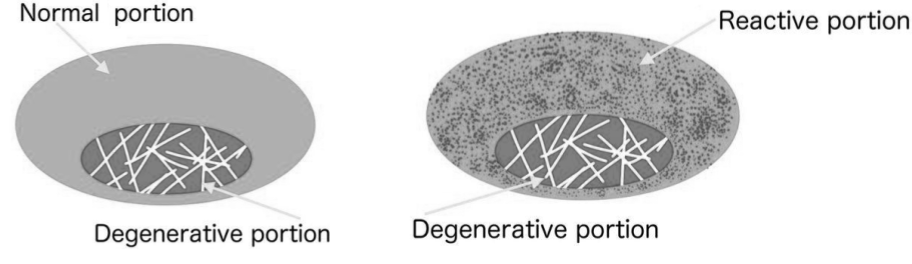
How Inflammation Affects Tendon Homeostasis
One of the hallmarks of inflammation is fluid-accumulation around the affected tissue. This is thought to be a mechanism to facilitate movement of immune-cells which are the ones who mediate inflammatory functions. Accumulation of fluid as well as an increased amount of active immune-cells in the peritendinous space are two characteristic histopathological findings in TP, and because of the delicate balance necessary to maintain nutrition in tendons, this fluid-accumulation and immune-activity threatens homeostasis if prolonged.52)https://pubmed.ncbi.nlm.nih.gov/31914656/53)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5714045/
Impaired Tissue-Nutrition
Contrary to how most people view them, tendons are not “passive” tissues when it comes to biomechanical load and similar to muscles they show increased expression of glycolytic metabolites such as lactate after exercise.54)https://www.ncbi.nlm.nih.gov/pubmed/1006691655)https://www.ncbi.nlm.nih.gov/pubmed/12382956 Tendons are a poorly vascularised tissue and the resident cell-population therefore needs to be adapted to function in a relatively hypoxic environment. Hence these cells already have a relatively increased reliance on non-oxidative metabolic pathways such as glycolysis, but one could predict that if oxygen-levels were decreased further there would be a reciprocal increase in the use of this metabolic pathway. So if the vascular exchange-mechanisms where to be compromised in a tendon we would expect to see increased level of glycolytic metabolites (as with any other tissue), both as a consequence of increasing levels of hypoxia and also because of impaired vascular export of lactate. In line with this studies have shown that increased levels of lactate and also other markers consistent with increasing hypoxia are present in TP.56)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2714139/57)https://pubmed.ncbi.nlm.nih.gov/20190320/
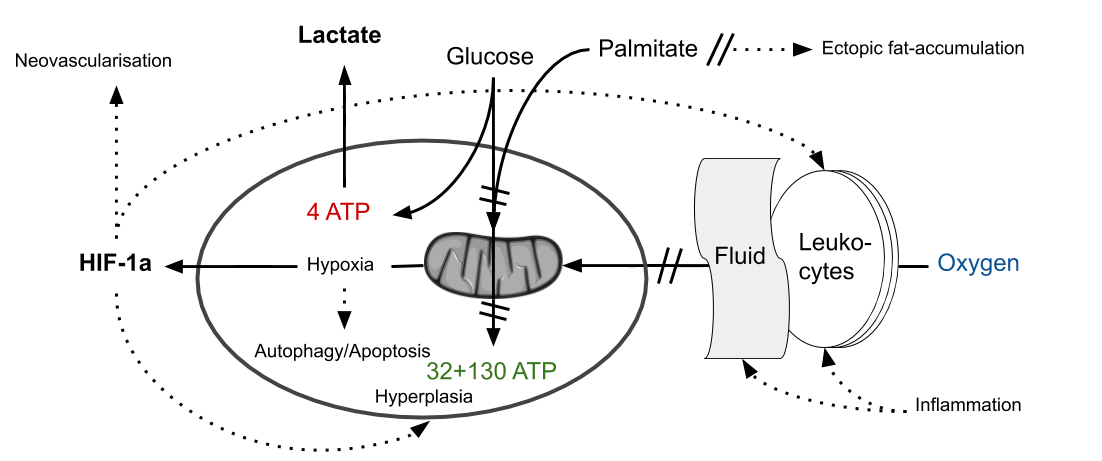
Justifying Neo-Vascularisation
An increasingly hypoxic environment could explain another histopathological characteristic of TP, which is the ingrowth of micro-vessels usually referred to as neovascularisation.58)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4650849/ This is commonly thought to be maladaptive but could perhaps be an attempt by the body to compensate for the effect of prolonged inflammation on local tissue-homeostasis.
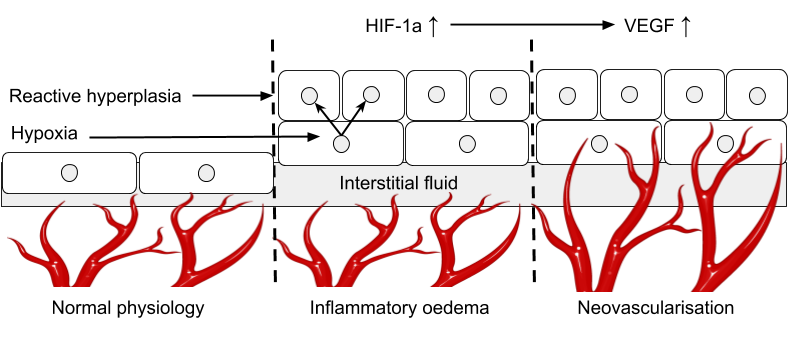
In a recent study, detectable levels of intratendinous blood-flow (a sign that the tissue has been infiltrated by neo-vessels, which in theory is pathological) was shown in 70-85% of runners with clinical TP, compared with 30-53% in controls with asymptomatic tendons at baseline, and following exercise an elevated level of blood-flow was seen in both groups, but it was severely prolonged in TP, which suggest two things. First, that tendons affected by TP in general seem to be in need of a greater vascular supply than healthy tendons, and second that it takes longer time for pathological tendons to recover their metabolic equilibrium after an insult, which could perhaps be because of a larger “oxygen-debt” in response to an anaerobically funded physical effort. Another study investigating vascular responses to exercise in tendons affected by TP have also showed that a failure to properly increase tendon-oxygenation in response to exercise was a significant risk-factor for TP prospectively in runners.59)https://pubmed.ncbi.nlm.nih.gov/29373799/ Together this suggests that neo-vessels could be serving a purpose, which perhaps could spare them form various methods of medical eradication such as sclerosing injections.
-More on the process of neovascularisation–
Inflammation as a Catalyst to Load-Related Injuries
Failure to meet the metabolic needs of the tendon during load would in theory predispose it to deterioration with increased intensity and frequency of mechanical load, and in this regard inflammation could be acting as a catalyst for TP by inhibiting nutritional exchange. In support of this, one study in mice showed that experimental induction of inflammation by injecting substance-P into healthy tendons lead to TP and that this effect was also accelerated by simultaneous application of mechanical load.60)https://pubmed.ncbi.nlm.nih.gov/33207770/ Another study also found that pharmacological inhibition of the inflammatory response completely abolished the catabolic response to exercise in tendons which also prevented the development of TP as a consequence of mechanical load.61)https://pubmed.ncbi.nlm.nih.gov/31560698/ A similar result has been shown after treatment with the anti-diabetic drug metformin, which has been shown to have anti-inflammatory properties (for reasons discussed in my article on nutrient-absorption and over-nutrition), and daily administration blocked TP-development with running-activity.62)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4990459/63)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7736509/
The Non-Mechanical Cause of Inflammation
From the previously mentioned studies, it is clear that mechanical load is involved in TP, but that it does not act alone in its pathogenesis and that other factors related to inflammation potentially constitutes the root-cause. Since the basis of the common theory of “micro-injuries” associated with over-use as the initiating factor is not supported by prospective studies, there is only one other thing that can drive inflammation while also flying under most medical radars, and that is a low-grade foreign/infectious threat (which is NOT the same thing as an actual infection).
Anti-Microbial Defence-Mechanisms are Activated in Tendinopathy
In support of such an etiology, studies have shown that diseased tendons show elevated levels of immune-receptors commonly associated with the anti-infectious response such as TLR4 and CD14.64)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5264298/65)https://www.ncbi.nlm.nih.gov/pubmed/29118051 Tenocytes taken från diseased tendons show an increased reactivity to the main microbial ligand of TLR4, bacterial lipopolysaccharide, and show robust up-regulation of inflammatory cytokines upon stimulation with this substance.66)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4883654/ Blocking TLR4-signalling in tenocytes taken from diseased tendons also seems to abolish the inflammatory activity inherent with TP.67)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5574425/
-More on the workings of inflammation in musculoskeletal disease-
A Compromise Between Tissue-Maintenance and Infectious Defence?
The immune-system has a dual role in the body as it is responsible for both repair and maintenance, and also defence against intruders. So analogous to a car repair-shop, if the mechanics are too busy keeping burglars out they wont be as good or as efficient at fixing cars resulting in half-finished repairs that fail to meet customer expectations, potentially also resulting in more “accidents”. There is plenty of evidence for a pivotal role of the immune-system in tendon-homeostasis, and one of the key-populations of cells here are the macrophages which have been shown to increase numerically in TP.68)https://www.ncbi.nlm.nih.gov/pubmed/32163543 69)https://onlinelibrary.wiley.com/doi/abs/10.1002/jor.24667 Macrophages are the most obvious example of cells playing dual roles, as in the case of tissue-damage or infection they turn into phagocytes that clears out debris and pathogens, while in the absence of such stimuli they instead adopt an anabolic phenotype where they starts to synthesise matrix-proteins as part of normal tissue-repair. Macrophage-phenotype is supposed to change according to the changing environment after injury, but in a study on tendon-injury in a diabetic mice it has been found that macrophages retain their phagocytic mode longer than controls and that this causes delayed healing and impaired mechanical properties which suggest that there is something interrupting the normal transition in macrophage phenotype in metabolic disease. 70)https://pubmed.ncbi.nlm.nih.gov/28686669/ There is currently insufficient evidence to conclude why, but my guess is that it is because of a low-grade infectious threat slowing or blocking this transition. In essence, that the “inflammatory environment” remains.
Evidence of Microbial Presence
It is currently not clear wether microbes are present in tendons affected by TP or if the inflammatory response is simply a reaction to microbial metabolites and/or endogenous danger-molecules circulating the vascular system, for which there is some evidence for.71)https://pubmed.ncbi.nlm.nih.gov/30728384/ Only one study has looked for direct evidence of microbial presence, and it confirmed that bacteria is present in ruptured achilles-tendons while being completely absent in control-tendons.72)https://www.ncbi.nlm.nih.gov/pubmed/28355086 However the technology used for detection in circumstances such as this when microbes might be present in very small numbers is still in its infancy and there is hence still great room for statistical errors in this kind of studies. Therefore, conclusions on this matter are precluded.
-More on microbial involvement in other chronic musculoskeletal diseases-
Connections With Diet
Studies have consistently shown that the same dietary model that cause obesity and DM2 also alters the mechanical properties in weight-bearing and non weight-bearing tendons in a manner consistent with TP.73)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6893090/ Interestingly, returning test-animals to a normal diet, the diseased tendons don’t return to their normal state despite normalisation of metabolic disturbances. This suggests that a metabolic insult can cause tendon-deterioration that is not sufficiently restored simply by removing the initial irritant which could perhaps explain the general (but limited) success of different loading-protocols which then presumably serves as a stimulus for an adaptive response to increase tissue-quality (assuming that the inflammatory irritant is no longer present). Tendons consist mostly of collagen, and one of the main amino-acids in collagen is glycine which is generally deficient in most modern diets, and there are data from a randomised trial that supplementation of glycine in the diet had superior effects when added to a loading-protocol compared with exercise alone.74)https://anatomypubs.onlinelibrary.wiley.com/doi/full/10.1002/ar.23041 Glycine-supplementation has also been shown to hasten recovery from experimentally induced inflammation in the achilles-tendon.75)https://pubmed.ncbi.nlm.nih.gov/25156668/ There are also other studies showing benefits beyond placebo for other dietary modifications and supplements in the treatment of TP.76)http://www.ncbi.nlm.nih.gov/pubmed/2692106677)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4915461/ Together this suggests that inadequate nutrition could be playing a part in the pathogenesis of TP.
Connections With the Gut-Microbiome
My guess however is that inflammation is the major factor that connects diet with TP, through diet altering the composition of the gut microbiome which then affects the infectious threat-assessment made by the immune-system causing an up-regulating of inflammatory defence-mechanisms.Though there are currently no studies that have directly tested this hypothesis specifically for TP, there are those that have shown an association between the state of the gut microbiome and tendon-homeostasis.78)https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0229908 The fact that gut micro-ecology could be playing a part in the pathophysiology of TP could also partly be supported by studies showing a strong association between TP and antibiotic-therapy (which could open up for intestinal colonisation with resistant microbes) with an almost 2-fold increase in risk for non-specific TP while the risk for TP in weight-bearing tendons such as the achilles is almost 4-fold compared to untreated controls.79)https://www.ncbi.nlm.nih.gov/pubmed/31270563 This could also be a direct consequence of a toxic effect, and interestingly, most substances used as antibiotics are in fact derived from microbial toxins.80)http://www.ncbi.nlm.nih.gov/pubmed/27477928 The most common way to induce experimental TP in animal-research is by injecting their tendons with collagenase, which is a toxin derived from microbes, and such microbes have also been shown to grow within the human gastro-intestinal tract, and many of them are associated with common infectious diseases in humans.81)https://pubmed.ncbi.nlm.nih.gov/24754251/82)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097604/83)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6624086/84)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC174012/pdf/641885.pdf Collagenase is no joke, and intravenous injection has proven to be fatal in mouse models.85)https://sci-hub.se/https://pubmed.ncbi.nlm.nih.gov/22197775/
-More on the microbiome, inflammation and their relationship with diet–
A Brief Note on Non-Steroid Anti-Inflammatory Drugs
Even though I argue here that inflammatory mechanisms are at the root of TP, that is not to say that directly inhibiting this mechanism, such as through common anti-inflammatory drugs is either wanted or beneficial. This is certainly not the case in rehabilitation, and in fact there is evidence to suggest that the stimulatory response that mechanical loading has with regards to tendon-healing and adaptation is impaired with such treatments.86)https://pubmed.ncbi.nlm.nih.gov/26159755/ A clinical trial also show that adding anti-inflammatory medications to physical rehabilitation does not provide any added benefit, and interestingly neither physical rehabilitation nor a combined rehab and medication-intervention show any structural improvement, which highlights that neither treatment-regime alter the significant prospective risk of clinical TP associated with structural TP, although they may both modify symptoms in the short term.87)https://pubmed.ncbi.nlm.nih.gov/33719579/
Understanding the Histopathological Picture
Histopathologically TP is a rather heterogenous condition, however there are more commonalities than differences between variants, and among the more universal characteristics of TP are the following:
- Increase in inflammatory mediators.88)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7006114/
- Loss of the normal collagen composition, density and architecture.89)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2505234/
- An increase in cell-numbers and a rounding of resident tenocytes.90)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5714045/
- Increased number of apoptotic cells and increased autophagy.91)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2505234/92)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2843163/93)https://pubmed.ncbi.nlm.nih.gov/19836974/
- Increased content of glycosaminoglycans and water.94)https://pubmed.ncbi.nlm.nih.gov/32882677/95)http://www.ncbi.nlm.nih.gov/pubmed/2731676196)http://www.ncbi.nlm.nih.gov/pubmed/17414481
- Hypoxia.97)https://pubmed.ncbi.nlm.nih.gov/31093655/98)https://pubmed.ncbi.nlm.nih.gov/20190320/
- Ingrowth of neo-vessels.99)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4650849/100)https://pubmed.ncbi.nlm.nih.gov/31093655/
Most of these features are commonly considered “maladaptive”, however I believe they can all be made sense of through the following narrative (take a deep breath!):
Injury or a pathogenic threat cause resident cells to send inflammatory signals and increase the synthesis of glycosaminoglycans. This serves to attract immune-cells to the exterior part of the tendon and increase the tissue water-fraction respectively. Fluid-accumulation and also a cytologically mediated change in the relative proportions of collagen-types and a re-organisation of collagenous geometry together form an antiseptic barrier and a scaffold for tissue-repair. This organisation aids the migration of tenocytes and immune-cells while also directly helping to clear debris and trap pathogens for removal. The combination of fluid-accumulation and increased immune-activity at the tissue-vascular interface impair the exchange of gases, nutrients and metabolites which cause hypoxia and an energy-crisis in the resident cell-population. Both the stimulus from hypoxic mediators and inflammatory irritants then cause a hyperplastic response in the resident cell-population, which serve both as a repair- and survival-mechanism as well as a defensive strategy. The increasingly hypoxic environment mimics the condition in cartilage whereby tenocytes start to drift towards an chondroid phenotype, which together with the increase in mitotic activity explain the rounder and more plump cell-shapes.101)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5738911/ The cellular energy-crisis cause an up-regulation of autophagy leading them to consume collagenous proteins to sustain homeostasis, which together with the possible contribution of microbial collagenases lead to a decrease in tissue collagen-content. To mitigate the negative effects of an impaired vascular exchange, neo-vessels try to penetrate into the tendon in an attempt to restore homeostasis, however this and the other histological reactions simultaneously compromises the mechanical integrity of the tendon and lowers its resilience towards tensile and compressive stress. If inflammation fails to resolve, or if neo-vascularisation and autophagy becomes insufficient to satisfy the homeostatic needs of the cells, they then become apoptotic and die causing a further deterioration of the tendon and its ability to adapt and maintain its resilience to load.
-More on the similarities in histopathology in chronic disease-
Understanding the Mechanical Consequences
As previously mentioned, histological change consistent with TP is not to be equated with the presence or the level of clinical symptoms. However, there are several objective mechanical consequences of TP that are present that put the individual at risk for injury, regardless of symptoms. What those mechanical consequences are seem to differ largely, both inter-individually and between different anatomical location.
Decrease Tendon-Stiffness
If the dominant histological change is more consistent with “regular” degenerative TP, then the tendon is softer and hence more compliant to load causing increased tissue-strain in response to the same level of pull. Data on the biomechanical tolerance of tendons show that strain-levels bellow 4% are physiologic and well tolerated while strain-levels that exceed this threshold result in micro-injuries, and if it exceeds 8% macroscopic failure is observed.102)https://pubmed.ncbi.nlm.nih.gov/16000201/ This is why tendon-stiffness has been shown to be dose-dependently related to load-tolerance.103)https://journals.physiology.org/doi/full/10.1152/japplphysiol.01449.2012
Increased Tendon-Stiffness
Not all tendons become softer with TP, and some actually become harder and hence less compliant to load compared with healthy tendons. Studies investigating the mechanical properties of tendons affected by TP have shown divergent patterns of stiffness in different anatomical regions, and while achilles-TP generally seems to be associated with decreased stiffness, patellar-TP show unchanged or increased stiffness compared with controls.104)https://onlinelibrary.wiley.com/doi/abs/10.1111/sms.12986105)https://link.springer.com/article/10.1007/s40279-018-0956-7 There also seems to be a difference in stiffness between cases of TP occurring within the same tendon in different individuals.106)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5738911/ In theory, increased tendon-stiffness could contribute to injury risk through decreasing the capacity for shock-absorption, causing elevated peak-loads in shock-absorption.
Reconciling the Differences
The reason for the above inconsistencies can be explained by differences is systemic pathophysiology, regional anatomy and local histology. The main difference between the patellar tendon and the achilles tendon is their cross-sectional area with the former being significantly larger than the latter. This difference means that nutrients have to traverse a longer distance to reach the inner portions of the patellar tendon which in the case of inflammation then predisposes to hypoxia in response to peripheral inflammation, and hence also to a phenomenon known as chondroid metaplasia. Chondroid metaplasia is the tendency of connective tissue-cells to assume a chondrocytic phenotype in response to changing environmental conditions, and the consequence is a partial replacement of tendon-tissue with cartilage. As the proportion of cartilage in a tendon increases its compliance to tensile load decreases resulting in a stiffer and less flexible tendon.
Advanced Glycation End-Products
There are studies that show that different metabolic disturbances cause different alterations to tendon-stiffness and experimental studies have shown that tendon-stiffening could be related to accumulation of something called advanced glycation end-products (AGEs). One such study showed that mice with a dysregulated lipid-metabolism fed a diet rich in AGEs developed stiffer tail-tendons compared with regular mice fed an obesogenic diet which instead developed softer tail-tendons.107)https://journals.physiology.org/doi/full/10.1152/japplphysiol.00584.2014 The main function of AGEs is thought to be to facilitate cellular adhesion (both for our immune-cells and microbes, depending on who’s making them!) and the mechanism by which AGEs act to stiffen tendons is thought to be by the formation of cross-links between collagen-fibers, decreasing inter-fibrillar sliding.
-More on advanced glycation end-products–
Insertional Tendinopathy
Stiffening of tendons poses a different kind of mechanical problem compared to tendon-softening in that while stiff tendons are more resilient to tensile load, they also increase the strain put on the muscle and the bony enthesis which could predispose to injuries at these locations.There is no data to support this assumption for muscle injuries, but there are evidence supporting that tendons affected by TP closer to the bony enthesis are associated with increased tendon-stiffness.108)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6056177/109)https://pubmed.ncbi.nlm.nih.gov/16858217/ What also differs the insertional part of the tendon from the rest is that it gradually transitions into cartilage followed by bone and hence if inflammation is centered close to the enthesis we might see a quicker increase in metaplastic cartilage progressing upwards along the tendons long-axis resulting in a stiffer rather than softer tendon as degeneration progress. As the proportion of cartilage in the distal end of the tendon increases, the tensile compliance decreases which can then increase the compressive load that the tendon is exposed to with motion in certain anatomical regions such as where the achilles-tendon passes over the calcaneal tubercle. The resulting pressure from motions such as end-range dorsiflexion could then amplify the local hypoxia while also adding a mechanical stimulus for adaptation of the tissue to compressive load.110)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6056177/111)https://pubmed.ncbi.nlm.nih.gov/16858217/ Both peritendinal bone-marrow oedema and sub-tendinal bursitis are both common MRI-findings in insertional TP.112)https://www.sciencedirect.com/science/article/pii/S235204772100023X#bib0085
Calcific Tendinopathy
Some cases of TP are associated with radiographic evidence of calcification which is most commonly observed in the tendons of the rotator-cuff (52-90% of cases) followed by the patellar tendon and the achilles tendon.113)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6209365/114)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3482552/
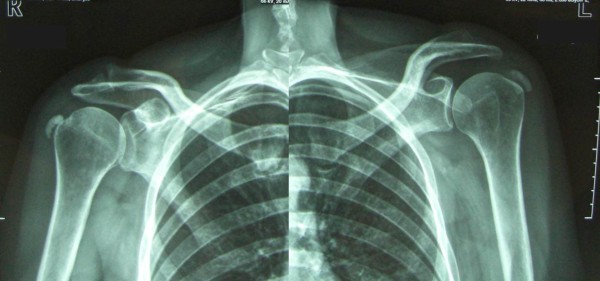
This phenomenon as well as formation of enthesophytes and other forms of extra-skeletal calcifications are most likely also a long-term consequences of hypoxia through a mechanism called endochondral ossification which naturally follow the process of chondroid metaplasia mentioned above.115)pubmed.ncbi.nlm.nih.gov/32293797 This is the same physiological mechanism by which most of our bones are formed in-utero, starting with a growing cartilage-model which grows to to the point where the radius to the center of the structure becomes too great to allow sufficient nutrition which cause hypoxia and apoptosis of chondrocytes, and a successive replacement with another tissue that is less dependent on an efficient vascular exchange, bone (note that because of its mechanical stability, bone can more easily harbour blood-vessels compared with cartilage).116)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7827519/
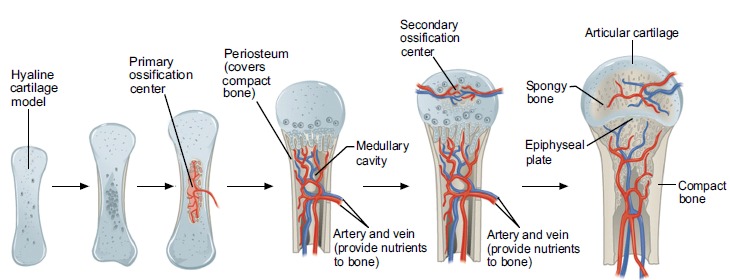
In line with this theory, tissues affected by ectopic calcification express a similar molecular signature as is expressed during endochondral bone-formation and the inhibition of signalling-pathways related to endochondral ossification halt the development of calcific TP.117)https://bmcmedicine.biomedcentral.com/articles/10.1186/1741-7015-10-95118)https://www.sciencedirect.com/science/article/pii/S001216060700245X119)https://pubs.acs.org/doi/abs/10.1021/acsbiomaterials.9b00335 Similarly as in endochondral bone-formation, the the calcified nodules also transitions radially into cartilage.120)https://sci-hub.se/https://doi.org/10.1093/rheumatology/41.4.474 Calcific TP has also been experimentally replicated by injection of collagenase into the central part of a tendon, showing that the condition does not have ties to systemic calcium-metabolism.121)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2628327/ Disturbances in normal convection as a consequence of reduced levels of mechanical load (perhaps as a side-effect of pain) might also play a part here, supported by data from in-vitro studies.122)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8142054/
A Metabolic Problem
Interestingly, endochondral ossification is also the mechanism that explain calcium-deposits in atherosclerotic arteries.123)https://pubmed.ncbi.nlm.nih.gov/31070451/ This explains why calcific-TP has been shown to be significantly associated with cardiometabolic disturbances such as diabetes, hypercholesterolemia and hypothyroidism, and when two or more of such disturbances are present at the same time the risk of developing calcific TP is 10-fold compared with cardio-metabolically healthy people.124)https://pubmed.ncbi.nlm.nih.gov/32976122/ Increased content of calcium-salts have been measured in degenerate tendons not affected by calcified TP and patients with diabetes also display elevated levels of calcium in their tendons compared with non-diabetic patients which suggests that calcific TP is simply a more severe form of TP and that it potentially has a systemic origin like previously discussed.125)https://pubmed.ncbi.nlm.nih.gov/8712860/126)https://pubmed.ncbi.nlm.nih.gov/2930276/127)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6209365/ This notion is further supported by the fact that patients with calcific TP also seems to be more commonly affected by other conditions characterised by ectopic accumulation of salt-crystals such as kidney-stones, gall-stones and gout.128)https://pubmed.ncbi.nlm.nih.gov/27047854/Together this suggests a systemic etiology for what on the surface seems to be a local condition.
Summary
From the evidence provided in this section, it is clear that the complexities of the painful tendon stretch beyond the biomechanical realm, and that it is likely rooted in metabolic pathology. However, since metabolic pathology itself is a multifactorial problem, and not only a question of diet/exercise, there are many other variables that have the potential to influence the origin and course of tendinopathy, but to this day they are still insufficiently studied. Looking for example at factors such as psychological distress, and the quantity and quality of sleep, which has been shown to be intimately involved with other conditions linked with chronic pain and metabolic diseases, there is no epidemiological or clinical evidence available at all for its involvement in tendinopathy. It simply hasn’t been studied. This is most likely because of the previous assumption that were dealing with a purely biomechanical problem, which has been commonplace for almost the entire time that the subject has been investigated, but this is successively changing with more and more research pointing to a systemic etiology, and less towards simple mechanical overload/underload.
To sum up, there are a few take-home messages I would like to underscore:
- First: inflammation is a primary feature of tendinopathy, not a secondary effect of injury, and its effect on homeostatic maintenance-mechanisms of cells/tissues looks to be the factor that blurs the relationship between wear and tear. When there is no interference or nutritional deficits, the tendons, like other bodily tissues, simply repair and adapt with changing levels of physical load (within reasonable limits!).
- Second: different variants of tendinopathy are simply different faces of the same problem, and they are all intimately tied to measures of metabolic health. Therefore, addressing the things that are interfering with metabolic health is essential to cause remission, in tendinopathy as with any chronic disease.
- Third: simply correcting or eliminating the systemic disturbance wont cause the tendon to regenerate, it will only stop it from degenerating, and although the pain may stop, it is crucial that potential nutritional issues are sorted out to enable sufficient collagen-synthesis, and physical rehabilitation should also probably still be carried out for a substantial length of time in order to lower the risk of rupture and reoccurrence.
In conclusion, tendinopathy is not the simple issue that it appears to be, which is evident both at the epidemiological, histopathological and clinical level. To reverse this condition, one has to get a good grip over many different aspects tied to general health, such as diet, sleep, physical activity and psychosocial wellbeing, in order to quench the workings of systemic inflammation. This is extremely difficult in practice, but unfortunately for those with any kind of chronic disease, there is no other way.
This is by no means all there is to say about tendinopathy, but this article has been laying in my pile of drafts for too long, and is starved for critical feedback from you readers, so please comment or email me if you have questions regarding this topic, or have spotted some errors.
Cover-picture by Alex Strachan
References