
Beyond Appetite
Nutrition in organisms is extraordinary complex, which is why it isn’t regulated solely by hedonic desire.
“Since growth and reproduction requires the accumulation of a nutrient-surplus by individual organisms, their continued existence depend on them being able to create and cope with scenarios of abundance, both behaviorally and physiologically. It is something that natural selection has wrestled with for aeons which has resulted in our present physiology for regulating nutrient ingestion and absorption. Overconsumption do present major problems to the individual, however their mechanisms and effects are better predicted by applying principles of ecology rather than toxicology”
Introduction
In modern society we tend to look at the biological world from a technical perspective, like a systems-engineer, and hence we often use artificial systems and machines as analogies to understand what’s happening in and around us, and why. In nutrition and dietetics, food is commonly considered to be a resource, analogous to fuel for a car, ignoring the vast ecological interplay that constitutes life and from this view-point metabolic diseases are often thought of as being states of nutrient toxicity, analogous to us accidentally but repeatedly overfilling the gas-tank because we’re not really sure of how much of it that can or needs to go in there. Excess gasoline then spills over into areas of the car where it is suddenly not a fuel anymore but a problem, such as to the vapor-collection system which eventually results in us having to visit the repair-shop prematurely. Modern petrol-pumps have a safety-switch that prevents us from making this mistake and we do as well, but since we are people, not cars, and that food not only fills our energy-stores but also makes us feel good we usually attribute the cause of metabolic disease to our hedonistic drives overriding this switch as a consequence of the palatability of the modern diet. Appetite is often thought of as being our only safety-switch and gate-keeper for nutrient intake and we hence assume that after food is ingested there is no-turning-back unless we trigger the gag-reflex. Nutrients dissolve into the blood and if we ate to much relative to our immediate needs or capacity for storage then the excess of nutrients such as fat and sugar spill over from the circulation into areas of the body where they shouldn’t be and cause direct toxic damage to cells, and later indirect damage through hemodynamic mechanisms such as in atherosclerotic cardiac hypoxia. Exactly like in the car-metaphor where excess gasoline ends up clogging the vapor-canister, which can cause what is called a vapor-lock1)https://www.moparmagazine.com/2018/07/too-much-in-the-tank/ that prevents fuel from reaching the engine.

However, there is a logical problem with using this model in dietetics. It doesn’t take into consideration the fact that the body has regulatory mechanisms for nutrient intake that goes beyond appetite, that other homeostatic control-functions are at work both pre- and postprandially before nutrients are ingested and before they pass from the gut into the blood. At the level of the small intestine, most nutrients are actually actively imported rather than passively absorbed, which means that the body could simply keep the doors shut on excess nutrients and let them pass through unabsorbed with stool, but does it, and if not then why?
In the following paragraphs of this section I’ve outlined how the body responds both histologically and physiologically to different states of nutrition and how this compares with what is seen in established metabolic disease. Through looking at how pre- and postprandial regulatory mechanisms are applied in these different states we then get to view the problem of over-eating from the perspective of the body. Is serum and tissue nutrient-toxicity really the main problem that the body is fighting when it comes to metabolic disease? Or is it something even bigger and more threatening?
In sum, this section aims to pick apart the axiom of direct tissue-damage through nutrient-toxicity as being the mechanism linking diet with pathology, and in the following section I’ll try to present what I believe to be a more probable hypothesis, that is also more coherent with fundamental biology and ecology to explain the connection between metabolic disease and over-eating.
Preprandial Histological Adaptations
Similar to how muscles adapt to physical load, other organs of the body also grow and decline to match their respective stressor accordingly, and the gastrointestinal tract is no exception.
Adaptation of Gastric Volume and Accommodation-Ability
Coming soon.
Adaptation of Small-Intestinal Mass
Studies have shown that an increase in nutrient intake leads to an increase in small-intestinal mass, and that this increase is because of a broadened absorptive surface through an increased number of enterocytes as well as an increased in the length and number of villi.2)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3806619/ It is in the interest of all living tings to grow and then when they reach a sufficient energy-surplus to reproduce, so this is likely part of a mechanism to rise above competition in acquisition of nutrients. The reverse is also seen with starvation, and in mice the intestinal wet-weight declines by almost 50% after just three days without food, but despite large losses of protein the over-all morphology and architecture of the intestine remains preserved.3)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2148066/ Like skeletal muscles, visceral tissues are costly to maintain and alone the small intestine accounts for about 20-35% of daily protein synthesis because of the continual cellular turnover that is necessary in the epithelial layer, and together with the other organs of the gut portal-system such as the stomach, spleen, liver and the pancreas they make up about a fifth of the total resting metabolic rate in the post-absorptive state while only representing less than 10 % of total body-mass, and when they are not needed they also serve as a tempting back-up reservoir of amino-acids that can also be converted to supply other tissues with glucose.4)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6334514/5)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2148066/ The negative effect of starvation on intestinal mass can be blocked by using leptin-infusion which signals sufficient lipid-stores, indicating that it is a last-resort to preserve energy-homeostasis.6)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6334514/ Contrastingly, in less severe forms of starvation such as caloric restriction test-animals have actually been shown to maintain, and even slightly increase intestinal weight and they also show a small increase in nutrient-assimilation.7)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4599246/
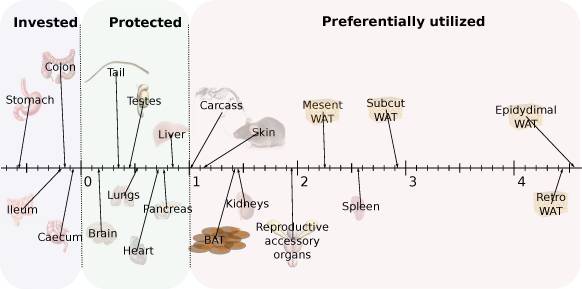
In relation to the small increase in intestinal mass, the capacity for nutrient-absorption is disproportionately enhanced in long-term caloric restriction because of an increased in nutrient transport-efficiency.8)https://pubmed.ncbi.nlm.nih.gov/9343490/ In one study, mice that were restricted to about 70% of ad-libitum food-intake over 270 days were shown to increase nutrient uptake-capacity by 20-100% and maintained intestinal mass similar to controls while instead sacrificing the mass of other internal organs such as the lungs, kidneys, spleen, heart, pancreas and liver which each differed by 40-120% compared with ad-libitum-fed controls. The same study also showed that mice subjected to acute total-starvation of one to ten days maintained nutrient absorption-capacity while at the same time displaying reduced intestinal mass per centimeter without altering the absorptive capacity per milligram of tissue, which suggest that they might manipulate gastrointestinal transit to accommodate this.9)https://pubmed.ncbi.nlm.nih.gov/11238759/ In experimental diabetes mellitus (DM), test-animals are seen to up-regulate intestinal nutrient absorption-capacity with almost a doubling of both total absorptive cell-numbers and intestinal wet-weight in long-term scenarios without a change in dietary composition.10)https://pubmed.ncbi.nlm.nih.gov/7633660/11)https://onlinelibrary.wiley.com/doi/abs/10.1002/jez.1401760410 Not by accident but through conscious effort. However, it is presently unclear how much of this is because of the hyperphagic response seen to occur after disease-manifestation as a consequence of tissue glucose-starvation, which have been shown to occur in animal-models of experimentally induced DM. This response is caused by increased synthesis and release of hunger-promoting hormones, and it can be blocked by replenishing liver-glycogen, which then interestingly also reduces hyperglycaemia.12)https://pubmed.ncbi.nlm.nih.gov/16484325/13)https://pubmed.ncbi.nlm.nih.gov/16484325/ It is also interesting to note that states of both over- and under-nutrition is met by an increase in intestinal mass and absorptive capacity, hinting that the body seem to be more concerned with the potential homeostatic threat of insufficient rather than excessive absorption. Not even in DM or obesity is there any indication that the body perceive serum over-nutrition as a threat, at least not as to warrant an adaptive decrease in absorption, so why is this?
Adaptation of Large-Intestinal Mass
Coming soon.
Adaptation of Intestinal Length
Comming soon.
Regulation of Nutrient Presentation to the Small Intestine
Since the absorptive capacity for most nutrients is close to zero in both the oral cavity, the oesophagus and also in the stomach, one of the main mechanism governing the rate of postprandial appearance of nutrients in the blood is the rate of gastric emptying into the small intestine where most of ingested nutrients are absorbed. This mechanism is in turn regulated through a complex interplay of different factors such as the contents of the diet, nutrient-status and sympathetic drive14)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6028327/15)https://pubmed.ncbi.nlm.nih.gov/7485502/ For the stomach to empty it usually takes one to four hours, which is quite a long time and hence endogenous control over the rate of gastric emptying has great potential to limit serum nutrient super-saturation in response to over-eating and in the following paragraphs we’ll explore how it responds to different nutritional states and how it is affected in metabolic disease.16)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4102158/
Gastric Emptying
Gastric emptying is most studied in relation to glucose-homeostasis and DM since it is the most significant factor that controls the postprandial appearance of glucose in the blood (also it accounts for about 35% of variance in peak postprandial glucose-concetration between individuals)17)https://pubmed.ncbi.nlm.nih.gov/25421372/ which in turn has long been thought to play a causal role in various metabolic diseases.18)https://www.sciencedirect.com/science/article/abs/pii/S1056872719306324 From the perspective of simple glucose homeostasis, the rate of gastric emptying seems to be involved as a regulatory mechanism, but with divided loyalties as studies have shown that it is affected separately by both serum and tissue glucose-status, which are not always in perfect harmony. In experimental settings increasing the level of glycemia cause a delay in gastric emptying implying that it works as a negative feedback-mechanism, but when hyperglycaemia is paired with tissue glucose-deprivation as in type-1 DM gastric emptying is instead consistently seen to increase.19)https://pubmed.ncbi.nlm.nih.gov/9207262/20)https://journals.physiology.org/doi/full/10.1152/ajpgi.00317.200721)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5917388/ In subjects with pre-diabetes defined as reduced oral glucose tolerance or increased fasting glucose-levels gastric emptying is shown to be delayed, implying saturated glucose-stores in these individuals, however in patient with manifest DM gastric emptying is instead shown to be normal.22)https://pubmed.ncbi.nlm.nih.gov/25725624/ In larger diabetic populations there does seem to be a significant portion of them that experience deviant patterns in gastric emptying and while still most of them have a normal rate, most of those who deviate from the norm show a delay while a lesser portion show an accelerated pattern, however the reason for these deviations are still not fully understood.23)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6028327/24)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6736218/ It is interesting to note that the dominant deviant pattern of gastric emptying in diabetic patients is a delay rather than an increase which would otherwise be predicted by the conventional view of DM progressing through high and frequent glycemic spikes in the blood postprandially.25)https://www.sciencedirect.com/science/article/abs/pii/S1056872719306324 In healthy subjects, longer gastric emptying-time has been shown to be positively related to satiety and also associated with lower caloric intake during meal-ingestion, which makes it an unlikely driver of metabolic states thought to be related to hyperphagia, and more likely a disease consequence.26)https://journals.physiology.org/doi/pdf/10.1152/ajpgi.00190.2017 Delayed gastric emptying in DM could be an attempt by the body to prevent postprandial glycemic spikes, however studies have shown it to be associated a greater risk of incident cardiovascular disease in DM which is the exact opposite of what one would predict from the postprandial glucotoxicity-model, and it also suggest that this is an impairment caused by the disease rather than an adaptation to hyperglycaemia, which is in line with early observations labelling it as “diabetic gastro-paresis” although the pathomechanics of this phenomenon has not yet been agreed upon.27)https://pubmed.ncbi.nlm.nih.gov/31177651/ However, contrary to popular opinion, delayed gastric emptying in DM doesn’t typically progress, and can also normalise with time.28)https://www.sciencedirect.com/science/article/abs/pii/S0168822719302906 Rates are also shown to be slightly increased in populations with well-controlled DM, and surprisingly it is most pronounced in patients treated with the anti-hyperglycaemic drug metformin.29)https://pubmed.ncbi.nlm.nih.gov/30933282/ Regardless, there does not seem to be any relationship between the rate of gastric emptying and diabetic phenotype, and deviant patterns are hence as just mentioned probably secondary consequence rather than causes or drivers of DM.30)https://pubmed.ncbi.nlm.nih.gov/18727706/31)https://pubmed.ncbi.nlm.nih.gov/31960554/ Although increased or decreased rate of gastric emptying does not seem to explain DM, postprandial glycemia does still seem to be a significant risk-factor for DM prospectively, however studies have shown that in those who later go on to develop the disease this elevation of postprandial glycemia at baseline doesn’t occur because of increased rate of glucose absorption or systemic insulin resistance, but instead because of impaired β-cell function which implies that this impairment could be preceding diabetic postprandial hyperglycaemia rather than that rapid spikes in glucose cause the disease.32)https://pubmed.ncbi.nlm.nih.gov/23539736/33)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6302905/
There doesn’t seem to be any consistent or significant relationship between gastric emptying-rates and obesity either, which together with the previous observations in clinical DM suggests that postprandial nutrient-delivery to the main absorptive organ of the gastrointestinal tract does not present a potent threat to serum-homeostasis, at least not so far as to warrant physiologic adaptations.34)https://pubmed.ncbi.nlm.nih.gov/22571886/
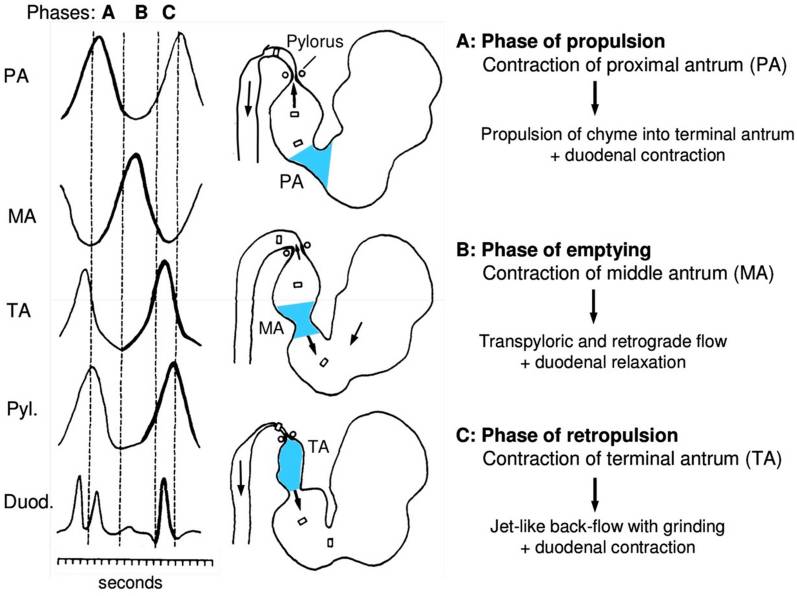
An important thing to consider with regards to potential preventive adaptation as to protect against serum nutrient-toxicity by adapting the gastric emptying-rate is that nutrients can’t simply leak into the duodenum but must be actively propelled onward, partly against gravity by muscular force as to be able to enter the small intestine. Because no absorption of the major macro-nutrients can take place in the stomach, there should not be any major direct physiological draw-back to simply hold on to gastric contents longer if the body wanted to avoid the potentially damaging effect of postprandial serum nutrient-overload (for those objecting at this point, please not that the “detergent effect” will be discussed in the coming section of this chapter). This would then also trigger earlier satiation and cessation of food-intake but this does not seem to occur, neither in obesity nor in established metabolic disease, at least not in the early stages. However, as previously mentioned in the pre-diabetic state there seem to be a decrease in gastric emptying-rate in some cases, and there are also studies that show an increased residual gastric volume in diabetic patients after an 8-hour fast, meaning that their stomachs have not completely emptied since they had their last meal, but the reason for this is still not clear.35)https://pubmed.ncbi.nlm.nih.gov/30609007/
Interestingly, and at first glance also a bit paradoxically is that gastric emptying rates have been show to be reduced in response to starvation, both in obese and lean subjects, and also in anorexia nervosa.36)https://pubmed.ncbi.nlm.nih.gov/7485502/37)https://bmcgastroenterol.biomedcentral.com/articles/10.1186/s12876-016-0560-y Why slow gastric emptying if there is a systemic nutrient-deficit, but not in diseases thought to be caused by to frequent/concentrated nutrient-delivery? One explanation could be that the body is more concerned with loosing nutrients than getting too much. In line with this, studies have shown that infusing nutrients into different portions of the small intestine cause different responses to gastric emptying and food-intake with an increased inhibitory effect on both in a proximal to distal fashion, meaning that gastric emptying and food-intake halts more the closer that infused nutrients get to the colon (this suppressing effect on food-intake is probably a major reason as to why gastric bypass works for weight-loss).38)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4808847/
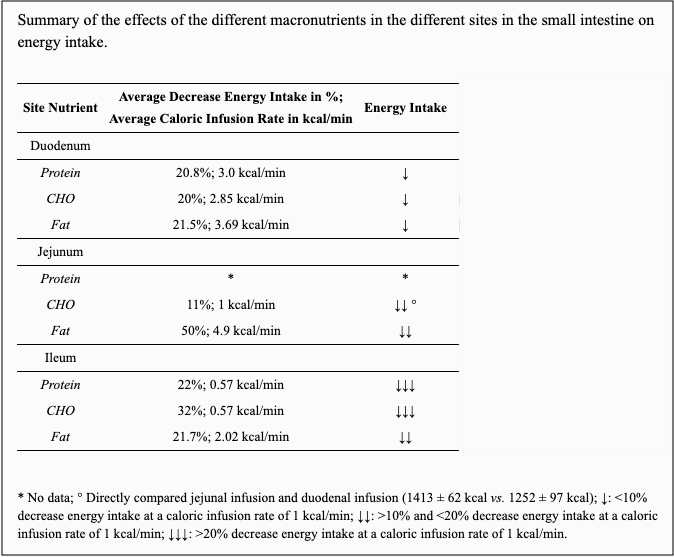
Studies investigating the difference between the suppressive effect of intra-venous and intra-intestinal nutrient-delivery on feeding-behaviour have also shown that the former did not have an effect but the latter did, which proves that this suppressive effect mentioned above is not because of increased levels of nutrients in the serum.39)https://pubmed.ncbi.nlm.nih.gov/8770012/ The reason for the proximal to distal difference in the inhibitory potential of nutrients on gastric emptying and food-intake is perhaps that there is a minimal capacity for absorption in the colon, and also about 1-2kg of hungry and potentially dangerous microbes there, so by not adapting gastrointestinal transit as to be able so sufficiently absorb ingested nutrients, the body risk both loosing nutrients through feces as well as being invaded by pathogens.40)https://www.sciencedirect.com/science/article/pii/S2352289516300509 If the body perceived acute postprandial serum nutrient-spikes as a major homeostatic threat then logically we would expect to see the reverse trend with higher or at least equally high inhibitory effect on gastric motility proximal to distal. Another interesting observation is that infusion of nutrients into the small intestine have different effects on food-intake depending on macronutrient-composition of infused bolus, and that there does not seem to be any significant difference in the magnitude of this suppressive effect between obese and lean individuals.41)https://academic.oup.com/ajcn/article/69/1/6/4694109
There is a strong negative correlation between age and gastric emptying-rates, meaning that if all other variables are equal we should be seeing lower postprandial glycemic scores with ageing, which is very far from the reality.42)https://pubmed.ncbi.nlm.nih.gov/26195094/43)https://pubmed.ncbi.nlm.nih.gov/30933282/
The most consistent relationship between nutrition and the rate of gastric emptying is meal-size and caloric content with higher numbers being associated with a longer emptying-time and lower numbers with shorter emptying-time (of note is that other dietary factors such as macronutrient-composition and osmolarity also play a big part). However, interestingly this positive relationship tends to plateau at meals containing three mega-joules after increasing strongly between meals of one mega-joule to two mega-joules.44)https://pubmed.ncbi.nlm.nih.gov/15150821/
To make sense of all these before mentioned findings perhaps we should consider gastric emptying to be mostly a mechanism serving to match delivery-rates with absorptive capacity (while absorptive capacity is matched with the needs of peripheral tissues), instead of viewing it as a mechanism working to preventing serum nutrient-overload. In support of this claim, gastric emptying-rates as well as the rate of intestinal absorption are seen to increase significantly within days/weeks after repeated large nutrient-challenges with the same nutrient.45)https://europepmc.org/article/pmc/pmc5371619
All the above observations are in line with the hypothesis that endogenous control over gastric emptying serves as a regulatory mechanism to maintain homeostasis and (perhaps) disregarding diabetic gastro-paresis, a delay in gastric emptying seems mainly to be a mechanism to spare nutrients both before and after they are eaten as not to overwhelm absorptive capacity and loose them to bacteria, and to a lesser extent delayed or increased gastric emptying may act as a regulatory mechanism that responds to tissue- and serum nutrient homeostasis. However, increased tissue- or serum-levels of nutrients in obesity and DM apparently don’t seem to present a sufficient homeostatic threat as to trigger the body to apply such mechanisms postprandially.
Regulation of Nutrient Absorption
Once ingested nutrients reach the first part of the small intestine, other regulatory mechanisms for absorption takes over. From here on, nutrient absorption is commonly thought to occur mostly as a passive phenomenon, that we take up nutrients like a sponge and that regulatory processes are activated first after nutrients have entered the bloodstream.46)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5609829/ The reality is that most absorptive processes, especially those of major macronutrients are heavily regulated and they also cost both energy and resources both to initiate and sustain. With this in mind, the nutrient-toxicity model makes very little biological sense, because it would infer that the body cause harm to itself, and not accidentally but consciously, analogous to fully instead of slightly opening the flood-gates when standing at the lower point of a dam when you’re thirsty, and knowing you’re going to drown, slowly.
The Exception of Water
This brings us naturally to water, which is the macronutrient that enters the body with the least amount of effort. Water-absorption occurs mostly through osmosis, however there are still active elements to the process which involves continual synthesis of aquaporins (the main cellular channels for water-absorption) and there are also other local regulatory processes put in place to adjust absorption-rate.47)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5037679/48)https://www.sciencedirect.com/science/article/pii/S2352304220300052 However, the level of regulation of a physiological process always has a reciprocal negative influence on its effectiveness, and since water needs to enter the body quickly in order to accommodate fluid-loss (which can occur very rapidly in large animals) and because of the large necessary daily turnover in terms of volume, the basal absorptive capacity needs to be very high and efficient. Hence the capacity of postprandial regulatory mechanisms are in this case very limited and ingestion of large amounts of water can cause fatal hyponatremia.49)https://www.ncbi.nlm.nih.gov/books/NBK537231/ This is presumably the reason why we seem to have more fine-tuned regulatory mechanisms for behaviour related to thirst than hunger. Disregarding cases of electrolyte-depletion, you seldom hear of someone who drank water (either neutral, sweetened or flavoured) to a point where the water had severe detrimental health effects just because they were really thirsty while obesity-related diseases clearly progress through mechanisms connected to appetite. However, nutrient deficiencies can definitely be a driver of obesity-related disorders, and this will be discussed in an upcoming section in this chapter.
Absorption of Glucose
Because of its role in diabetes, glucose was long though to be absorbed from the gut through simple diffusion. However there were never any real evidence for this and it has now been known for over a decade that glucose crosses from the intestinal lumen to the blood almost exclusively through an active transporter known as Sodium-Glucose Co-Transporter 1 (SGLT1) that are expressed on the apical membrane of enterocytes which uses energy to create a sodium gradient that it then uses to import glucose into the cell.50)https://journals.physiology.org/doi/full/10.1152/physrev.00055.2009 Assisted, or facilitated diffusion does occur through a transport-protein known as Glucose Transporter 2 (GLUT2) but its contribution is minimal to total absorption even in the context of high-carbohydrate diets and its level of expression is also fairly context-independent, hence it is non-essential. In contrast, point-mutations in the genes that controls for SGLT1 have proven to be fatal by causing severe diarrhoea (not through starvation) but only in the context of a high carbohydrate diet and mice without functional SGLT1 thrive normally when glucose and galactose is either entirely absent or present in lower levels in the diet.51)https://pubmed.ncbi.nlm.nih.gov/20385036/52)https://pubmed.ncbi.nlm.nih.gov/20385036/53)https://europepmc.org/article/pmc/pmc6516085#free-full-text54)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3237647/ The concentration of SGLT1 on the apical membrane of enterocytes increases and decreases in response to varying levels of glucose in the diet and in the blood (in contrast to GLUT2 which remains fairly constant), and also varies diurnally to match anticipated intake according to dietary habits.55)https://www.ncbi.nlm.nih.gov/pubmed/1533370656)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2749303/57)https://www.tandfonline.com/doi/abs/10.1080/09168451.2018.1451743?journalCode=tbbb20 Interestingly, SGLT1 also increases in response to ingestion of artificial sweeteners meaning that the mechanism perhaps is reinforced through respondent conditioning.58)https://www.physiology.org/doi/full/10.1152/ajpgi.00068.2016 Many have argued that intestinal sugar-absorption is unregulated, but there are several emerging lines of evidence available now that supports the existence of negative feedback-loops.59)https://www.jbc.org/content/273/23/14545.long60)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7462918/61)https://www.nature.com/articles/labinvest2015129
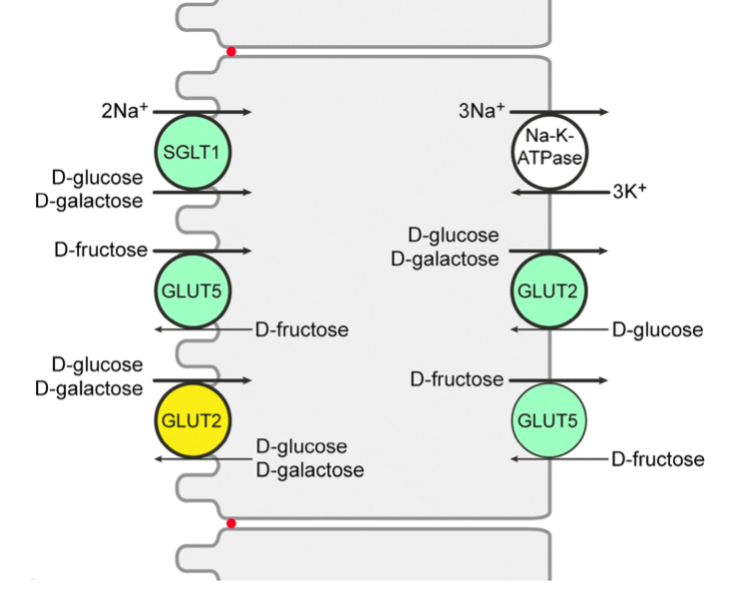
Studies in piglets consuming iso-caloric diets have shown that basal levels of SGLT-1 are sufficient to absorb dietary glucose as long as the total carbohydrate-content of meals do not exceed 50%, which means that intestinal absorption is the first rate-limiting step in response to high carbohydrate loads.62)http://dx.doi.org/10.1017/S0007114510000954 Paradoxically (from the conventional perspective), when the mechanism that increases intestinal SGLT-1 in response to increased levels of dietary carbohydrate is absent, postprandial levels of glycemia rises and test-subjects also show an abnormal insulin-profile in response to oral glucose-loads.63)https://doi.org/10.1017/S0029665111000103 The reason as to why SGLT-1 aren’t present at high levels on enterocytes all of the time is because they are very costly to maintain. SGLT1 itself doesn’t use energy, but to function it needs Na/K-ATPase to be co-expressed at the enterocyte basal membrane which in turn uses energy to create a chemical gradient of sodium which then enables glucose to move against its concentration-gradient through SGLT1 by hitchhiking with sodium-ions from the intestinal lumen.64)https://link.springer.com/article/10.1007/s00424-020-02439-5/ If no glucose is present this mechanism is just an expensive energy-leak, but unlike a lamp connected to a light-switch this mechanism can’t simply be turned off when its not needed so instead the body, analogously, needs to break either the lightbulb or the light-switch and make a new one when illumination is needed again. Since SGLT1 and Na/K-ATPase are proteins this affects nitrogen-balance and a study has shown that protein-restriction causes reduced levels of SGLT1 and serum-glucose which implies that dietary and/or endogenous protein is directly needed and used for glucose transport.65)https://academic.oup.com/jas/article-abstract/90/13/4995/4703514 So if maintaining a high glucose absorption-capacity actually requires increased use of both energy and precious nitrogen, it seems very strange that the detrimental health effects of excess dietary glucose are blamed on hyperglycaemia. Paradoxically (again, from the conventional perspective), diabetic patients and patients with obesity have increased levels of SGLT1 in their intestines compared to healthy and normal-weight controls and elevated levels of SGLT1 also seems to occur as a response to starvation in animal-studies.66)https://academic.oup.com/jn/article/130/9/2174/468654367)https://journals.physiology.org/doi/full/10.1152/ajpgi.00068.2016 There is also data showing an increase in glucose absorption-rate by about 70% when test-animals were fed a calorie-restricted diet.68)https://pubmed.ncbi.nlm.nih.gov/1021087/ This reinforces the hypothesis that the homeostatic control-mechanisms are directed from signals of nutrient-deficiency in the diabetic state rather than responding to a toxic serum nutrient-excess, and that this activates glucose-sparing strategies contrary to anti-glucotoxic mechanisms. This is further supported by data showing that the increased levels of intestinal SGLT1 (and GLUT2 and GLUT5) in DM are reversed by insulin-therapy probably by giving a false signal of a shift from a catabolic state to an anabolic state.69)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7462918/ The anti-hyperglycaemic drug metformin that is commonly used in management of DM2 has paradoxically been shown to causes an increase in intestinal expression of SGLT1 and increased glucose-absorption in the intestines of patients with the disease.70)https://www.sciencedirect.com/science/article/abs/pii/0006295295022430?via%3Dihub71)https://www.sciencedirect.com/science/article/pii/S016882271730356X Metformin originates from a class of toxins that are used by some plants as a defence-mechanism as not to be eaten by herbivores, and an interesting observation related to oral metformin-treatment is that up to 300 times higher concentrations of the drug accumulate in the intestine than in the circulation. The underlying mechanism of effect of the drug seems to be that it causes an increase in intestinal glucose-metabolism by up-regulating the level of glycolytic activity in enterocytes, which reaches such a high level that it also causes these cells to consume additional glucose from the systemic circulation, this then creates a glucose deficit, which in healthy individuals causes an up-regulation of endogenous glucose production in the liver, but in the diabetic state the reverse happens with a decrease in endogenous glucose production.72)https://pubmed.ncbi.nlm.nih.gov/32994049/73)https://pubmed.ncbi.nlm.nih.gov/28776081/74)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946719/ This may also be because DM is a form of starvation at a physiological level, and that the process that synthesises glucose de-novo is very metabolically expensive. With regards to the effects of metformin on glucose-metabolism, it is also interesting to consider that the body actually has the capacity to burn off excess glucose in the diabetic state but that it still chooses not to do so. It is also interesting to note that DM is often seen to increase both in prevalence and severity with advancing age, however, enterocyte absorption-capacity of glucose as well as other nutrients are instead seen to decline.75)https://pubmed.ncbi.nlm.nih.gov/8447410/76)https://pubmed.ncbi.nlm.nih.gov/8463853/
Absorption of Fructose
Like with glucose, absorption of fructose is also regulated postprandially by differential expression of its respective transport-protein; GLUT5 (and to a lesser extent; GLUT2) which are allocated to the apical membrane of enterocytes as to match levels of dietary intake.77)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6457363/ Several studies has shown GLUT5 to be the dominant transporter and genetic deletion causes a 75 % drop in small intestine fructose-absorption on a high-fructose diet, and similarly to what happens with SGLT1-inhibition, impairing GLUT5 is fatal in experimental models, but only in the context of a high-fructose diet .78)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4550372/79)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2643499/ After binding to GLUT5, fructose then enters the enterocyte by facilitated diffusion, but even though the main motive force in this mechanism is passively mediated by a concentration-gradient, fructose-absorption is not to be mistaken for a passive process. The transport-proteins required for fructose uptake are present only at very low levels in the basal state, in fact they are almost non-existent in most species of mammals from birth to the end of the weening-stage, and up to that point the rates are almost identical in both omnivores, carnivores and herbivores and they only diverge after fructose-containing foods are differentially introduced to the diet. 80)https://journals.physiology.org/doi/abs/10.1152/ajpgi.1992.263.5.G60581)https://journals.physiology.org/doi/abs/10.1152/ajpgi.1992.263.5.G59382)https://journals.physiology.org/doi/abs/10.1152/ajpgi.1990.259.4.G54483)https://www.annualreviews.org/doi/10.1146/annurev.ph.51.030189.00312584)https://journals.physiology.org/doi/full/10.1152/physiolgenomics.00056.2004 Dietary levels of fructose actually seems to be the only signal which can modulate intestinal GLUT5-expression, and mice that are gavaged with fructose show a rapid 6-fold increase compared to when on their regular diet. This means that they are only at about 17% of their maximal capacity in the normal state. Over time, dietary patterns then establishes a diurnal rhythm that’s independent of the vagus nerve and hence work through local intestinal signalling pathways and/or via endocrine mechanisms.85)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2652499/ The fact that intestinal GLUT5 varies with dietary intake of its substrate tells us that like with SGLT1 there might be costs (or dangers) involved in maintaining the transporter in the absence of the substrate that outweighs the benefits of having a more passive and unregulated system of absorption. If portal or systemic fructose could reach toxic levels simply through dietary over-consumption, that would pose a great problem for frugivorous animals because of the palatability of fruits in the setting of intermittent availability and the delay in appetite-regulation in relation to serum and tissue nutrient-status. Same as with SGLT1, because GLUT5 is a protein and that the enterocytes in the intestinal lining are renewed every few days, a high level of continual synthesis of this protein is required to maintain a high absorptive capacity which similarly affects nitrogen balance, and like SGLT1 the intestinal expression of GLUT5 are also elevated in patients with type-II diabetes.86)https://journals.physiology.org/doi/full/10.1152/ajpgi.00310.2001 So if chronic fructose-poisoning is thought to play a causal role in metabolic disease linked with insulin-resistance through post-absorptive mechanisms, it then seems paradoxical that maintaining a high fructose absorption-capacity requires post-prandial effort by the body, and that this is also sustained sufficiently long as to cause problems. Interestingly though, and contrastingly with SGLT1, intestinal GLUT5 actually seems to be lower in patients with obesity compared with normal-weight subjects.87)https://pubmed.ncbi.nlm.nih.gov/28936585/ Because very little fructose escapes the portal system, where the liver turns most of dietary fructose into glycogen and fat for storage, only traces of fructose make it to the systemic circulation. Hence intestinal absorption of fructose might be negatively regulated in part by the level of saturation of hepatic energy-stores that then inhibits intestinal GLUT5-expression, implying that there might be a negative feedback-mechanism in place. The contrast between obese patients and those with DM2 with regards to intestinal GLUT5 could then perhaps be explained by impaired glycogen storage mechanisms in the latter, which have been confirmed in animal-studies of experimental DM2.88)https://pubmed.ncbi.nlm.nih.gov/2145494/ Interestingly, like with SGLT1, the levels of intestinal GLUT5 is also increased rather than decreased in response to the anti-diabetic drug metformin.89)https://www.sciencedirect.com/science/article/abs/pii/0006295295022430?via%3Dihub
Absorption of Lipids
Postprandial regulation of the appearance-rate of lipids in the serum is also governed by transport-proteins at the enterocyte-level, and like in the previous two examples they are also adjusted to match dietary intake. Fatty acids enter into enterocytes mainly by endocytosis through the assistance of membrane-structures knowns as caveolae which are flask-shaped invaginations in the plasma-membrane. Caveolae are a subdomain of what is called lipid-rafts which are micro-domains in cellular membranes enriched in sphingolipids and cholesterol that mediates cellular trafficking-mechanisms, such as endocytosis.90)https://www.sciencedirect.com/science/article/pii/S193764481082003991)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3179338/ In addition to being simple lipid-rafts, the caveolae are also enriched with specific proteins called caveolins (CAVs) which are the key molecules that mediates intestinal fat-absorption.
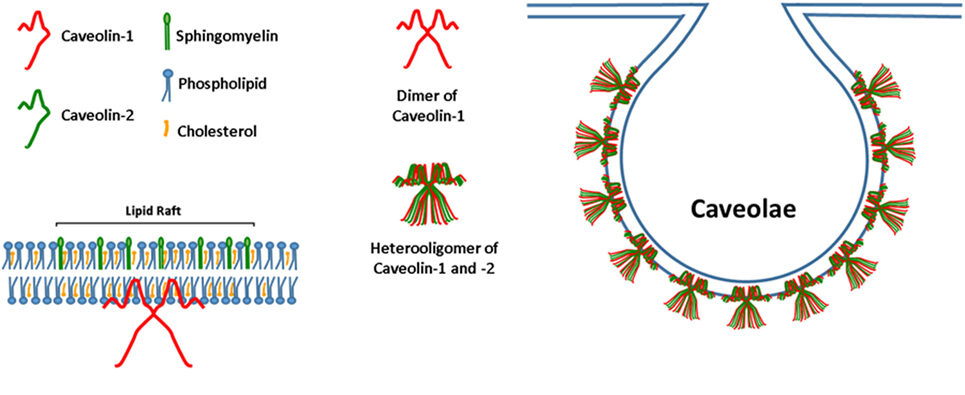
The motive force in trans-membrane migration of dietary fat are like fructose mainly driven by a concentration gradient, i.e. through diffusion, but this is only enabled through continual synthesis of cholesterol, sphingolipids and CAVs.92)https://www.sciencedirect.com/science/article/pii/S1937644810820039 Other membrane-associated proteins also play a role in intestinal lipid absorption, they include cluster determinant 36 (CD36), plasma-membrane associated fatty acid-binding Protein (FABPpm) and a family of fatty acid transport proteins 1-6 (FATP1-6). They can collectively be referred to as fatty acid-binding proteins (FABP).93)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3996833/ However, most of these FABP mainly serve regulatory functions related to post-absorptive processes that takes place after the lipids have entered into the enterocyte, such as the formation of chylomicrons. In line with this, studies that experimentally blocked FABP signalling-pathways have not shown any significant effects on absorptive capacity from the intestinal lumen while deletion of the genes coding for CAVs such as the CAV-1 gene have been shown to cause steatorrhea in response to a high-fat diet.94)https://pubmed.ncbi.nlm.nih.gov/23958439/95)https://www.sciencedirect.com/science/article/abs/pii/S0300908413002812?via%3Dihub Another very interesting observation seen in a recent study on mice is that deletion of the CAV-1 gene cause an increase in serum cholesterol and free fatty-acids as well as systemic insulin resistance and decreased glucose-tolerance in response to a high-fat diet despite a reduction in the gain of total body-fat and visceral fat-mass compared with wild-type mice.96)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7432291/ Some of these effects was also seen in response to the control diet when compared with wild-type mice which is in line with other studies who saw a similar metabolic phenotypes in adipocytes taken from wild-type mice fed a high-fat diet and from CAV-1 mutant mice fed a normal diet.97)https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0046242 However, hyperlipidemia in mutant mice lacking CAV-1 can partly be explained by the fact that other cells of the body such as adipocytes and endothelial cells are also partly dependant on a functioning CAV-1 gene for absorbing lipids which could mean that there is indeed a problem emerging as a consequence of lipolysis and back-flow from end-organs that cause serum-supersaturation with lipids which in turn causes them to spill over into the artery-walls.98)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3149783/ This hypothesis can actually be quickly dismissed as studies using selective inhibition of enterocyte CAV-1 also show increased serum levels of fat and cholesterol, and that both local and global inhibition of CAV-1 actually confers resistance to diet-induced atherogenesis which means that lipids in atherosclerotic plaques are also actively recruited and imported rather than passively deposited into “places where they shouldn’t be”.99)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5374320/ Atherogenesis in cardiovascular disease might actually be a repair-response secondary to vascular injury and in line with this a study has shown that CAV-1 mutant mice fail to regenerate their blood-vessels after ischemic injury.100)https://pubmed.ncbi.nlm.nih.gov/15205364/ Interestingly, CAV-1 mutations carry with them a long list of severe detrimental health-effects, and among those is vascular dysfunction as well as pulmonary hypertension, right ventricular hypertrophy and cardiomyopathy.101)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2291498/ In other words: blocking intestinal fat-absorption blocks atherosclerosis, yet this seems to cause the very diseases that were originally thought to be caused by to much fat being absorbed and deposited into the artery-walls. In humans, CAV-1 mutations are also seen to cause sever lipodystrophy and progeria.102)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4503302/ Another very interesting observation is that CAV-1 expression has been shown to be increased in diabetes and when pharmacologically blocked this has been shown to cause co-morbid alzheimers-disease.103)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7007939/104)https://www.jneurosci.org/content/39/43/8576 There is also evidence to suggest that that intestinal fat-absorption is regulated through negative feedback as mice who lack the intestinal receptor for leptin, which is a hormone that is synthesised by adipocytes when they approach their storage-limit, display 28% increase in fecal lipids and a delayed onset of obesity compared with normal mice when consuming an obesogenic diet while no difference is observed when both groups of mice consumed a control-diet.105)https://faseb.onlinelibrary.wiley.com/doi/pdf/10.1096/fj.14-255158
Absorption of Cholesterol
Cholesterol absorption is also mediated by endocytosis and is also regulated postprandially by a transportprotein known as Niemann-Pick C1 like 1 (NPC1L1). The intestinal expression of this protein has been shown to be increased in clinical and experimental diabetes and the causal link is unsurprisingly thought to be hypercholesterolemia.106)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3543648/ Also, studies have shown that disabling NPC1L1 cause a dramatic decrease in cholesterol absorption which also blocks the development of atherosclerosis which has put a large target on its back.107)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3465994/108)https://pubmed.ncbi.nlm.nih.gov/11742881/ Cholesterol is an essential component of cell-membranes and steroid-hormones, and the debate about the cause of hypercholesterolemia has mostly been about if it is a pathological phenomenon related to excess dietary intake or endogenous synthesis, and if the latter is the case whether this occurs as response to stress and/or injury or if it is an aberrant response. Essentially: does the body want and use more cholesterol in metabolic disease, or is hypercholesterolemia a problem of endogenous production or dietary intake exceeding serum-clearance? A clue to the potential answer in view of the perspective that has been brought forth in this section is that in hyperlipidemic men, statins which are the most widely used drug in pharmacological treatment of hypercholesterolemia through down-regulation of endogenous synthesis has been shown to increase NPC1L1-expression in the intestine which does indeed seem to suggest that the body needs more cholesterol, not less into the system in metabolic disease.109)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3035692/
Absorption of Peptides and Amino-Acids
Protein is perhaps the most controversial macronutrient in dietetics, but their potential role in metabolic disease is not as frequently discussed as that of sugars and fats. In the intestinal lumen, proteins are broken down into peptides and amino-acids, and they are then imported into the enterocytes by active ion-dependent transport-mechanisms mediated by transport-proteins from the solute carrier (SLC) and cadherin (CDH) gene-families, assisted by ATP-consuming ion-exchangers that are involved in maintaining the desired chemical gradient that enables transport. 110)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5872740/ The level of expression of these proteins and the capacity for absorption varies with dietary protein-content.111)https://pubmed.ncbi.nlm.nih.gov/7911522/ Like with many sugars and fats, there is also strong evidence for a correlation between increased serum-levels of certain amino-acids and metabolic diseases such as type-II diabetes, and the level of expression of peptide-transporters such as SLC15A1 are increased compared to healthy controls.112)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4839172/113)https://www.nature.com/articles/s41598-019-43431-z114)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6359471/115)https://pubmed.ncbi.nlm.nih.gov/29239245/116)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5726373/ Interestingly, the levels of SCL15A1 are also increased in response to fasting and starvation, and pharmacological attempts at inhibiting SLC15A1 instead led to increased expression of this protein, meaning that they are very hard to silence.117)https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5726373/ This reason might be tied to the essential role that proteins play in other metabolic processes which in turn might be the main drivers of increased intestinal expression of these transporters, like the fact discussed earlier that protein-metabolism is intimately intertwined with to glucose-metabolism. An interesting finding is also that complete silencing of SLC15A1 in the upper small intestine, when successful have been shown to disrupt glucose-homeostasis in response to a high-protein diet.118)https://www.nature.com/articles/s41467-018-03490-8
Adjustments to Intestinal Transit-Time
Coming soon.
Age-Related Changes in Absorptive Capacity
Most metabolic diseases and their consequences are seen to increase in prevalence and incidence with increasing age. However, an interesting observation is that nutrient-absorption-capacity does not follow this pattern, and is instead shown to decrease with advancing age which also seems quite contradictory to conventional etiological models.119)https://pubmed.ncbi.nlm.nih.gov/8463853/
The Paradox of Nutrient-Absorption in Metabolic Disease
Since potential nutrient availability to organisms has always varied throughout biological history, sometimes being over-abundant and sometimes entirely absent, it would not make sense for there not to be some kind of feedback-mechanism that controls postprandial nutrient-absorption as to prevent excess nutrients from disrupting homeostasis. Especially since there is an unavoidable lag between appetite, feeding behaviour and nutrient-status. Indeed, there seem to be such mechanisms at work, but they instead seem to further enable and enhance, rather than limit absorption of nutrients in obesity and in metabolic disease, which suggest that over-nutrition is not really the type of problem that we imagine it to be for the body.
To disallow nutrient-entry to the circulation the body doesn’t really have to do anything, only stop doing. The main paradox here thus is that if direct tissue gluco-, fructo-, lipo- or amino-toxicity is thought to be the main-mechanism in which over-eating causes disease, then why doesn’t the body try to limit nutrient-absorption instead of actively enhancing it in the context of metabolic disease? In DM, fatigue of pancreatic beta-cells caused by hyperglycaemia is usually blamed for inducing a state of glucose-starvation, which may cause some people to liken these responses to gasping for air while being submerged under water. But the question stated in the introduction then still remains: why doesn’t the body just let excess nutrients pass out with the stool in the initial stage of over-nutrition before metabolic disease ensues if it already has enough energy and are slowly taking damage from serum nutrient-overflow? Especially if it saves nitrogen in the process, which is much harder to come by in ecosystems relative to energy-substrates such as glucose.
If we start out with the assumption that the body is an intelligent organism moulded by millions of years of evolution the facts presented in this section when taken together instead seems to suggest that metabolic disease are states of deprivation at a physiological level rather than a states of over-abundance, and an intriguing thought is that they might also be states of enhanced (or failed) adaptation to competition for nutrients with other organisms. From this perspective we may then consider if the association between metabolic diseases and over-eating isn’t directly causal, but that it might instead potentially be mediated through mechanisms related to other organisms such as the intestinal microbiome acting as a third variable in these complex dietetical equations.
Over-Nutrition Cause a Eco-Biological Problem Rather Than a Direct Biochemical Problem
Matching nutrient-ingestion with the needs of the body while also succeeding to seize opportunities for growth and reproduction are the fundamental goals of all organisms, and inter-and intra-species competition for these goals are also what maintains all of biology. Since growth and reproduction requires the accumulation of a nutrient-surplus by individual organisms, their continued existence depend on them being able to create and cope with scenarios of abundance, both behaviourally and physiologically. It is something that natural selection has wrestled with for aeons and has for now resulted in our present physiology for regulating nutrient ingestion and absorption. Overconsumption do present major problems to the individual, however their mechanisms and effects are better predicted by applying principles of ecology rather than toxicology. Hypothetically, in both small- and large-scale ecological scenarios if one or a few organism grew powerful enough to seize control of all others and then used them as nutrients to enable their own reproduction, biodiversity would disappear and life would fall apart. The reason why this scenario hasn’t occurred (yet..) is because of one simple fact: it’s easy to rob someone who holds more than he can carry. So luckily at the right time in evolution emerged the practice of parasitism, which balances out the ecological playing-field such as that if one organism accumulates too much, someone els could just come in and skim of the excess and hence get a competitive advantage. In this way, too much success breeds parasites.
“There is a problem that comes with a life characterised by abundance, but it only becomes visible when we look at the situation through an ecological lens. When an organism manages to acquire valuable resources at a rate that exceeds its immediate need and capacity for efficient storage, they become increasingly attractive targets for parasites, wether it be large ones such as foxes and squirrels, or microscopic ones such as bacteria and fungi.”
The act of eating something is not like charging your phone-battery, it is an interaction with other living organisms and as I’ve argued for in the Model-chapter of this book, I believe that disease in general can be said to be phenotypic expressions of one-sided (parasitic) ecological interactions, with most cases occurring between a host-organism and its commensal microbes, at least in the context of modern human life. As will be outlined in the next section, when the will of other organisms is included in dietetical equations, the relationship between over-eating and metabolic disease becomes too complicated to be explained by a direct nutri-toxic effect. Also, if other ecological players such as microbes are indeed the causal link between over-eating and disease, then it could mean that processes related to nutrient ingestion and absorption are not unanimously controlled by the host, and might be influenced by other organisms, further complicating the subject on pre- and postprandial regulation of nutrient absorption that have been discussed in this section.
If the theories put forward here are correct, then this puts the causal mechanism of metabolic disease more within the realm of ecology and biology rather that pure biochemistry and toxicology, and perhaps there we can make sense of how the body handles states of over-nutrition..
References